








In situ structure determination of virus capsids imaged within cell nuclei by correlative light and cryo-electron tomography
Posted on: 3 May 2020 , updated on: 25 May 2020
Preprint posted on 6 February 2020
Article now published in Scientific Reports at http://dx.doi.org/10.1038/s41598-020-74104-x
A close, really close glimpse into the world of viruses within the host cell using innovative Cryo-EM
Selected by Debakshi MullickCategories: biophysics, microbiology, pathology
Background
Cryogenic electron microscopy (Cryo-EM) has recently emerged as a reliable method to determine high-resolution biomolecular structures of a range of samples, from proteins, nucleic acids, and viruses to whole cells. Unlike in a conventional transmission electron microscope (TEM), the microscope and the samples are maintained at cryogenic temperatures (~-190°C). The samples are prepared by plunge-freezing them in a bath of liquid nitrogen-cooled ethane on a TEM grid. The water freezes rapidly to a disordered glassy state (vitrification) [1], without forming crystalline ice that would both cause damage and diffract strongly on encountering the electron beam, thereby obscuring the sample.
Cryo-EM is often a preferred choice where dehydration, harsh fixatives, and metal contrasting agents alter the native structures and give rise to undesirable artefacts e.g. determination of high-resolution 3D structures of purified proteins. For larger samples like viruses (or for cellular ultrastructure), cryo-electron tomography (cryo-ET) is preferred where a series of 2D TEM projections are obtained by tilting the sample through ~120° and are computationally back-projected to obtain the 3D structure. The resulting tomograms can be used to build high-resolution structures in a method called sub-tomogram averaging.
The current work focuses on the pathogenic herpes simplex virus-1 (HSV-1), which is a causative agent of oral herpes or ‘cold sores’. HSV-1 is a fairly large-sized virus (~1300 Å) that initially replicates within host nuclei to give rise to newly-formed virions whose capsids now enclose the dsDNA genome of the virus [2]. In the cytoplasm, they are enclosed in a protein-rich layer (the tegument) that is made up of assemblies called capsid-associated tegument complex (CATC), which is composed of 3 main proteins viz pUL17, pUL25, and pUL36. Here, Vijayakrishnan et al. aimed to elucidate the infectious cycle of HSV-1 within the host cell using Correlative Light Electron Microscopy (CLEM) to identify foci of replicating virus in a confocal microscope, before cryo-ET of these regions. The authors successfully integrated several EM and analysis techniques to shed more light on the in situ infectious cycle of HSV-1.
Results
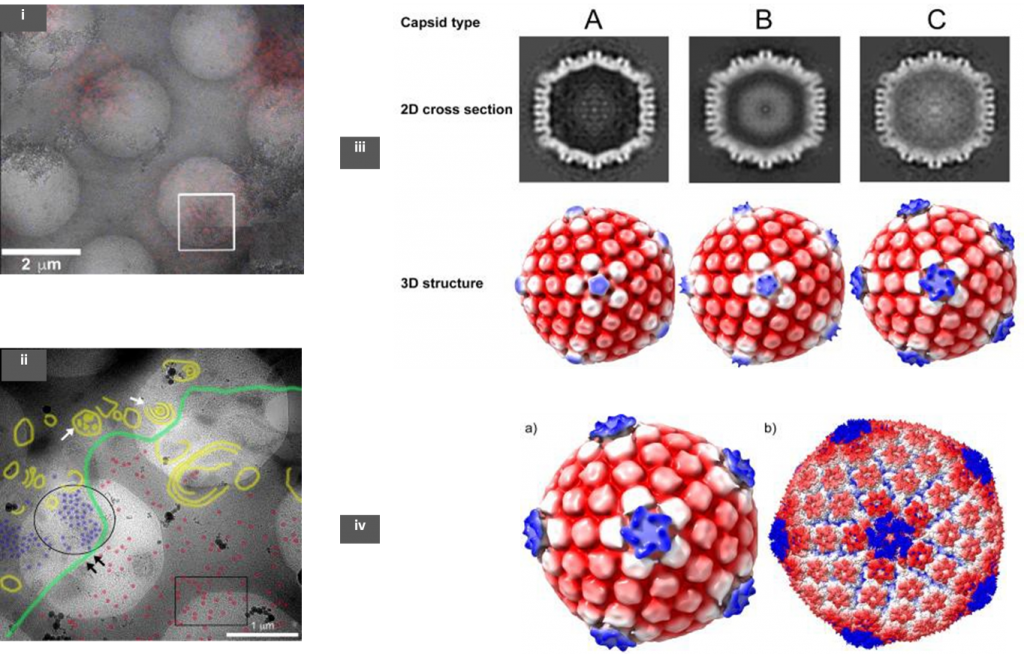
The authors used an RFP tagged HSV-1 to infect the host baby hamster kidney cells (BHK-21). Samples were prepared using the Tokuyasu method, in which the infected cells were fixed with aldehydes and embedded in gelatin before being frozen in liquid nitrogen [3]. Following this, sections of ~200 nm were cut using a cryo-microtome and collected on a holey TEM grid. These sections were observed under a confocal microscope to identify RFP expressing HSV-1 regions, before cryo-ET of these regions of interest (ROIs) (Fig 1; (i)). These grids were then re-vitrified by plunging in liquid ethane and low magnification TEM images were then overlaid on the fluorescence map (CLEM). Tomograms were obtained in regions that showed a good density of viruses.
The precise sub-cellular (nucleus or cytoplasm) recruitment location of pUL36 to CATC of the virus has been of a debate in the field. To lay this to rest, the authors have used a mutant strain of the virus- FRΔUL37 (null mutant for pUL37 tegument protein) to infect host cells. The pUL36 protein is vital for virion particles to bud off from the nucleus whereas the absence of pUL37 does not any adverse effect on nuclear egress. Unlike the wild-type strain, HSV-1 FRΔUL37 nucleocapsids are not directed to the trans-Golgi network and accumulate in the cytoplasm [4]. Cryo-ET was done at both cytoplasmic and nuclear locations where there was a large density of virion particles (Fig 1; (ii)).
Three kinds of capsids were observed from cryo-ET- empty (A), scaffold-protein containing (B), and DNA filled (C), whose structures were later determined from sub-tomogram averaging (Fig 1; (iii)). It is noteworthy to observe that both B and C capsids were seen in the nucleus but the cytoplasm contained mostly C-capsids suggesting that the virion was fully formed along with the CATC before nuclear egress. Structural analysis of the 3 types revealed that only C-capsid had similar CATC densities as seen in previous averaged data from purified HSV- 1 [2,5]. The calculated resolution obtained from this analysis was about 6.4nm (Fig 1; (iv)). The observed CATC densities on the C-capsid were further corroborated using a focused classification method that can further refine structures within an already averaged structure.
In summary, the data emerging from the analysis adequately points in the direction that pUL36 recruitment happens within the nucleus as almost all C-type nucleocapsids have CATC densities that are comparable to a fully mature virus. This is the first time that precise structures have been determined from viruses that are within the nucleus- the virus’s replication site.
Comments and questions
See the author’s response section
Why I chose this article
I have an interest in cryo-EM of samples in their functional context i.e. within whole cells. This preprint effectively integrates several aspects of cryo-EM very innovatively. The work-flow is simple to follow and it is promising for those who want to study cellular and macromolecular ultrastructure in their near-native form. The methods do not entail the use of highly specialized instruments making it plausible for many labs to be able to adopt it straight away.
References
- Dubochet, J., Adrian, M., Chang, J., Homo, J., Lepault, J., McDowall, A., & Schultz, P. (1988). Cryo-electron microscopy of vitrified specimens. Quarterly Reviews of Biophysics, 21(2), 129-228. DOI: 10.1017/S0033583500004297. Cryo-electron microscopy of vitrified specimens. Rev. Biophys. 21, 129–228 (1988).
- Dai, & Zhou, Z. H. Structure of the herpes simplex virus 1 capsid with associated tegument protein complexes. Science (80-. ). 360, eaao7298 (2018).
- Tokuyasu, K. T. Immunochemistry on ultrathin frozen sections. J. 12, 381– 403 (1980).
- Pasdeloup, D. et al. Inner tegument protein pUL37 of herpes simplex virus type 1 is involved in directing capsids to the trans-Golgi network for envelopment. Gen. Virol. 91, 2145–2151 (2010).
- McElwee, M., Vijayakrishnan, S., Rixon, F. & Bhella, D. Structure of the herpes simplex virus portal-vertex. PLOS Biol. 16, e2006191 (2018).
doi: https://doi.org/10.1242/prelights.19875
Read preprint (3 votes)
(3 votes) Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the biophysics category:
Mechanically-induced Septin Networks Protect Nuclear Integrity
Filipe Nunes Vicente
Loss of Sun2 ablates nuclear mechanosensing-driven extracellular matrix production and mitigates lung fibrosis
Beth Chopak
Shape independent fluidisation in epithelial monolayers
Sindhu Muthukrishnan
Also in the microbiology category:
Circadian Clock Programming of Anticipatory Antiviral Immunity Gates Enteric Virus Infection Susceptibility
Owen Ang
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
Gut microbiome changes over the course of multiple sclerosis differentially influence autoimmune neuroinflammation
Carole Djagang et al.
Also in the pathology category:
Behavioral characteristics of an extremely old rhesus macaque in a zoo: Dementia-like symptoms and implications for quality of life of geriatric animals
Stefan Friedrich Wirth
EBV reprograms autoreactive anti-CNS B cells as antigen presenting cells in multiple sclerosis
Léa Bastien et al.
Clinically reported covert cerebrovascular disease and risk of neurological disease: a whole-population cohort of 367,988 people using natural language processing
Rafidah Mumtahinah Chowdhury et al.
preLists in the biophysics category:
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
preLights peer support – preprints of interest
This is a preprint repository to organise the preprints and preLights covered through the 'preLights peer support' initiative.
| List by | preLights peer support |
66th Biophysical Society Annual Meeting, 2022
Preprints presented at the 66th BPS Annual Meeting, Feb 19 - 23, 2022 (The below list is not exhaustive and the preprints are listed in no particular order.)
| List by | Soni Mohapatra |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Biophysical Society Meeting 2020
Some preprints presented at the Biophysical Society Meeting 2020 in San Diego, USA.
| List by | Tessa Sinnige |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Biomolecular NMR
Preprints related to the application and development of biomolecular NMR spectroscopy
| List by | Reid Alderson |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |
Also in the microbiology category:
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
BioMalPar XVI: Biology and Pathology of the Malaria Parasite
[under construction] Preprints presented at the (fully virtual) EMBL BioMalPar XVI, 17-18 May 2020 #emblmalaria
| List by | Dey Lab, Samantha Seah |
1
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the pathology category:
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
COVID-19 / SARS-CoV-2 preprints
List of important preprints dealing with the ongoing coronavirus outbreak. See http://covidpreprints.com for additional resources and timeline, and https://connect.biorxiv.org/relate/content/181 for full list of bioRxiv and medRxiv preprints on this topic
| List by | Dey Lab, Zhang-He Goh |
1
Cellular metabolism
A curated list of preprints related to cellular metabolism at Biorxiv by Pablo Ranea Robles from the Prelights community. Special interest on lipid metabolism, peroxisomes and mitochondria.
| List by | Pablo Ranea Robles |