








Molecularly distinct models of zebrafish Myc-induced B cell leukemia
Posted on: 3 December 2018
Preprint posted on 6 November 2018
Article now published in Leukemia at http://dx.doi.org/10.1038/s41375-018-0328-1
ALL together: development and comparison of distinct zebrafish models of B cell Acute Lymphoblastic Leukemia
Selected by Hannah BrunsdonCategories: cancer biology
Background
Acute lymphoblastic leukemia (ALL) arises from lymphoblasts within the bone marrow. Lymphoblasts differentiate into either immature T or B cells, which are the cells-of-origin for the two main subclasses of ALL, T-ALL and B-ALL. Zebrafish models of T-ALL have been used successfully for the past 15 years, as their haematological, immunological and oncological pathways are highly conserved in humans. However, despite the clinical relevance of pre (precursor)-B-ALL, as it is the most common paediatric cancer, zebrafish B-ALL models went unrecognised until very recently.
Earlier this year, the Langenau and Frazer labs both published independent reports in the journal Leukemia detailing their discovery of zebrafish B-ALL models within pre-existing T-ALL models [1],[2]. Both groups used zebrafish expressing an oncogenic Myc transgene driven by the B/T cell-specific recombination gene rag2 promoter, which caused fish to develop ALLs. It was assumed from previous research that only T-ALL leukemias would develop in these models, but unexpectedly, both groups found a subpopulation of leukemias that were phenotypically and transcriptionally characteristic of B-ALL, or in one case, a mixture of B- and T-ALL (termed biphenotypic ALL). Where the two models differ is the species origin of the Myc transgene, and the genetic background of the fish – the Langenau group use a murine-derived Myc transgene (mMyc) and the Frazer group use a human-derived transgene (hMYC). Although these transgenes are highly similar, it raises the possibility that the leukemias generated by the two models might have important differences that could both undermine or expand their usefulness in modelling human ALL.
In this preprint, the two groups show the results of their collaboration to validate, compare and contrast these B-ALL models at the transcriptional level. Happily, they confirm both models as driving B-ALL formation alongside T-ALL, and also find that unexpectedly, their B-ALLs originate from different B cell lineages, thus potentially modelling different subtypes of B-ALL.
Key findings
First of all, the authors performed RNA-seq on 13 hMYC ALLs so that they could be compared to the published mMyc RNA-seq data. Principal component analyses of expression profiles from both datasets showed that the data clustered not according to species’ transgene, but whether they were more characteristic of T-ALL or B-ALLs, with the single biphenotypic ALL clustering between the two groups, as shown in Figure 1A from the preprint, below:
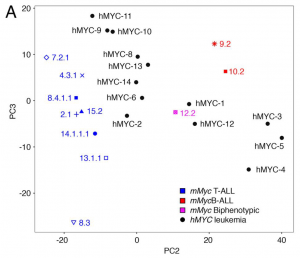
From Figure 1A of the preprint (under CC-BY-NC-ND 4.0).
This is a PCA plot of RNAseq expression profiles from previously classified mMyc-induced ALL (blue, red and pink), and hMYC-induced ALL (black). The previously proposed hMYC T-ALLs (2, 6, 8-11, 13,14) cluster near mMyc T-ALLs (blue), proposed hMYC B-ALLs (3-5) near mMyc B-ALLs (red) and two hMYC ALLs (1 & 12) near the mMyc biphenotypic-ALL (pink).
The identities of tumours within these clusters was confirmed by hierarchical clustering with the top 100 positively/negatively regulated genes, which showed that all T-ALLs expressed the same T cell markers, B-ALLs expressed B cell markers and biphenotypic/mixed ALLs expressed a mixture of these.
Differences between the two models were revealed upon investigating the clonality and maturation of B and T cell receptors in hMYC and mMyc ALLs. Perhaps unsurprisingly, the T cell receptor tcrβ was expressed in all T- and biphenotypic-ALLs, but absent in B-ALLs. Interestingly, B-ALL leukemias differed in which B-cell receptor they preferred to express, with hMYC B-ALLs favouring the ighz form and mMyc-driven B-ALLs favouring the ighm form. Ontological comparison of genes uniquely expressed in T-ALLs, hMYC ighz+ B-ALLs and mMyc ighm+ B-ALLs, confirmed a transcriptional divergence between the two B-ALL types.
Taken together, this suggests that although the oncogene driving B-ALL in both models is very similar, they appear to be derived from different B cell lineages. This is exciting, as it means that we have gone from being able to model just T-ALL in zebrafish, to being able to model three more subtypes of ALL – hMYC ighz+ B-ALL, mMyc ighm+ B-ALL, as well as biphenotypic ALL.
Why I chose this preprint
The ‘Confirmatory Results’ is a new tag for me on Biorxiv.org, as most of the uploaded reports I’ve found so far fall into the ‘New Results’ category. With the rise in media coverage about the lack of reproducibility in scientific studies, it is good to see groups collaborating to confirm and build upon their research, and to make these findings accessible to everyone. From a disease modelling standpoint, detailed characterisation and comparison of new models is important so we know how closely they recapitulate different facets of human pathologies. It will be exciting to see how these B-ALL models can now be employed to explore any differences in their molecular aetiologies and treatment responses alongside their T-ALL counterparts.
Questions
- It is intriguing that hMYC and mMyc transgenes give rise to transcriptionally divergent B-ALLs – are there any differences in tumour latency, aggressiveness and phenotype in general that could arise from these transcriptional differences?
- Do the different transcriptional profiles of ighm+ and ighz+ B-ALLs correlate with those arising from particular B cell lineages in patients?
- What are the key questions you want to address first with your B-ALL models?
The articles this preprint compares:
Borga, C. et al., 2018. Simultaneous B and T cell acute lymphoblastic leukemias in zebrafish driven by transgenic MYC: implications for oncogenesis and lymphopoiesis. Leukemia.
Garcia, E.G. et al., 2018. Cell of origin dictates aggression and stem cell number in acute lymphoblastic leukemia. Leukemia, 32(8), pp.1860–1865.
doi: https://doi.org/10.1242/prelights.6127
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the cancer biology category:
Profilin 1 maintains cell cycle fidelity to prevent unscheduled genome doubling and polyploidy in cancer
Akshata Samel
Aging increases ovarian cancer growth, metastasis, and immunosuppression that can be alleviated by inhibiting hedgehog signaling
Zoha Sadaqat
A pre-rRNA positive feedback loop drives malignant ribosome biogenesis
Vaishali Grewal
preLists in the cancer biology category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Anticancer agents: Discovery and clinical use
Preprints that describe the discovery of anticancer agents and their clinical use. Includes both small molecules and macromolecules like biologics.
| List by | Zhang-He Goh |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |