








Ribosome rescue inhibitors clear Neisseria gonorrhoeae in vivo using a new mechanism
Posted on: 18 June 2020 , updated on: 19 June 2020
Preprint posted on 5 June 2020
Lost in trans-translation: acylaminooxadiazoles target Neisseria gonorrhoeae by blocking the rescue pathway of non-stop ribosomes
Selected by Zhang-He GohCategories: microbiology, pharmacology and toxicology
Background of preprint
Neisseria gonorrhoeae, the pathogen responsible for the sexually transmitted infection (STI) gonorrhoea, has a high global public health burden. Despite the formation of the World Health Organisation (WHO) Global Gonococcal Antimicrobial Surveillance Programme (GASP), the global surveillance of gonococcal antimicrobial resistance remains limited in many regions [1]. Worryingly, N. gonorrhoeae has developed resistance to a range of antibiotics, such as sulfonamides, penicillins, tetracyclines, macrolides, fluoroquinolones, and early-generation cephalosporins [1]. As a result, ceftriaxone is being used in many countries as the sole remaining empiric monotherapy for gonorrhoea [1].
To fill the urgent need for alternative mechanisms of action in the treatment of gonorrhoea, Aron et al. target the bacterial ribosome rescue pathway, a trans-translation pathway that is unique to bacteria and has no mammalian equivalent [2]. In previous experiments involving E. coli (Fig. 1), the Keiler group found that acylaminooxadiazole derivatives inhibit trans-translation by binding the ribosome [3]. In this work, the authors develop their initial hit, KKL-35, developing it as a potential antibiotic against N. gonorrhoeae (Fig. 1).
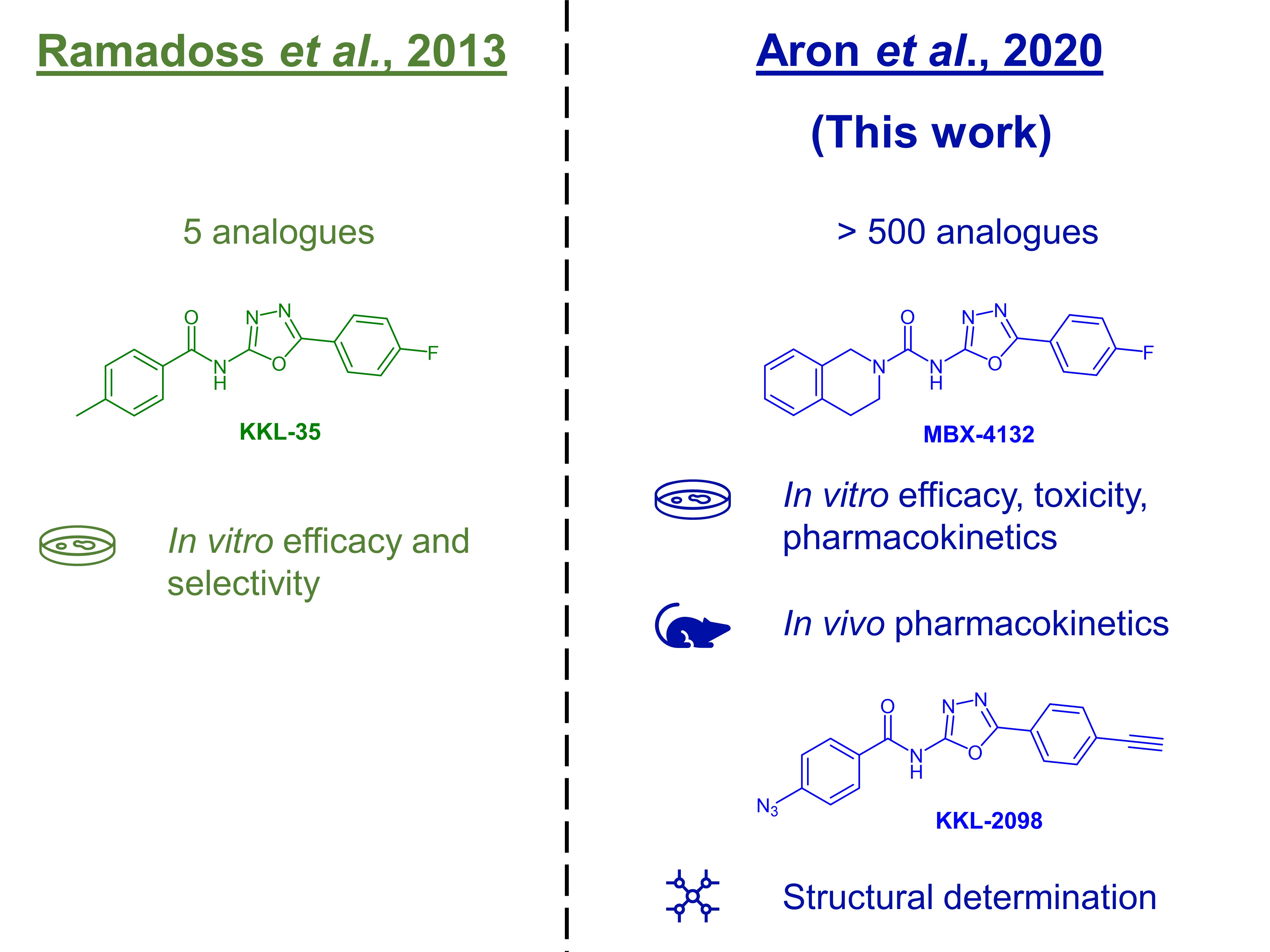
Figure 1. The initial discovery of KKL-35 and its subsequent development.
Key findings of preprint
Because the amide bond in KKL-35 was easily hydrolysed in initial in vitro pharmacokinetic studies, Aron et al. determined that KKL-35 was unsuitable for animal studies. The authors then synthesised over 500 analogues, leading them to identify the structure-activity relationship (SAR) among these compounds (preprint Fig. 1A, preprint Table S1). This process led Aron et al. to discover MBX-4132.
In their preprint, the authors discuss several considerations in determining whether MBX-4132 will be a clinically viable antibiotic: pharmacokinetics, efficacy, and toxicity.
(A) Pharmacokinetics
The authors found that MBX-4132 exhibited desirable pharmacokinetics both in vitro and in vivo. MBX-4132 had good metabolic stability (in vitro murine liver microsome assay) and permeability (Caco-2 assay). In mice, MBX-4132 was also highly bioavailable (Fsc = 34%, FPO = 46%), exhibited a long half-life (t1/2 = 3.55 h), and had a low clearance rate (CL = 72.6 mL/hr/kg).
(B) Antimicrobial activity
Aron et al. then described several aspects of MBX-4132 that describe its antimicrobial activity. First, not only was MBX-4132 broadly active against Gram-positive and Gram-negative species, but it was also effective against clinical isolates of N. gonorrhoeae—including MDR strains—which showed that MBX-4132 could circumvent most existing resistance mechanisms. Second, the authors found that there was a low frequency of spontaneous mutants resistant to MBX-4132, leading them to conclude that emergence of clinical resistance was likely to be slow. Third, time-kill assays revealed that MBX-4132 was bactericidal, rather than just bacteriostatic, at concentrations at ≥ 4x minimum inhibitory concentration (MIC). Fourth, MBX-4132 was also effective in vivo: a single oral dose completely cleared 80% of mice of infection by 6 days, accompanied by a significant reduction in bacterial load (preprint Fig. 2).
The authors also confirmed the mechanism of action of acylaminooxadiazole antibiotics, using cryogenic electron microscopy (cryo-EM) to determine the structure of KKL-2098 (a related acylaminooxadiazole analogue) cross-linked to its target (preprint Fig. 3).
(C) Toxicity
In their preprint, the authors also described three key experiments that suggest that MBX-4132 is likely to exhibit low toxicity in mammals. First, Aron et al. conducted several in vitro assays to show that MBX-4132 displayed minimal off-target activity: competitive binding with mammalian receptors and cardiac ion channels; inhibition of major liver CYP450 enzymes; and an Ames assay. Second, MBX-4132 did not significantly affect the mitochondrial membrane polarity in human hepatocytes in vitro. Third, the in vivo pharmacokinetic assays, MBX-4132 also did not seem to generate clear adverse effects in mice.
What I like about this preprint
This preprint was a particularly good example of how drug discovery is done. The authors previously developed a hit [3], and this preprint shows how they then further developed it using further preclinical studies.
I liked how the authors addressed the two main considerations in pharmaceutical development: safety and efficacy. To understand these issues commonly faced by pharmaceutical experts, the authors considered not just the pharmacodynamics (preprint Fig. 3) but also the pharmacokinetics of MBX-4132. Both in vitro and in vivo experiments were conducted in an extensive series of experiments to prove both points. To answer the safety question, the authors conducted experiments ranging from mutation tests such as the Ames assay, to in vitro selectivity tests with HeLa cells, to in vivo pharmacokinetic tests in mice. To answer the efficacy question, the authors conducted in vitro tests on clinical isolates as well as in vivo tests in a murine genital tract infection model.
There are two other clinical points to consider in the continued development of MBX-4132 as well. First, significant drug-drug interactions may occur when one drug is significantly metabolised by, or inhibits, a given enzyme. In this case, the authors did not identify any significant inhibition of major CYP450 metabolising enzymes by this MBX-4132 (preprint Table S5), which reduces the risk of drug-drug interactions. Second, there is also the issue of clinical dosing. Given the relatively long half life of these compounds and the bactericidal concentration-dependent mode of action of MBX-4132, it may benefit from reduced dosing—or even a single oral high dose, as the authors demonstrated in vivo. Taken together, these two clinical implications augur well for the clinical utility of MBX-4132.
Future work
As Aron et al. suggest, most future work will likely involve determining the mechanism of action of MBX-4132 in greater detail. Gaining a better understanding of the exact nature of the SAR of this class of acylaminooxadiazole ribosome inhibitors will help to refine the rational design and development of these molecules.
References
[1] Wi T, Lahra MM, Ndowa F, Bala M, Dillon J-AR, Ramon-Pardo P, Eremin SR, Bolan G, Unemo M, Antimicrobial resistance in Neisseria gonorrhoeae: Global surveillance and a call for international collaborative action, PLOS Medicine 14(7) (2017) e1002344.
[2] Ramadoss NS, Alumasa JN, Cheng L, Wang Y, Li S, Chambers BS, Chang H, Chatterjee AK, Brinker A, Engels IH, Keiler KC, Small molecule inhibitors of trans-translation have broad-spectrum antibiotic activity, Proc Natl Acad Sci U S A 110(25) (2013) 10282-10287.
[3] Alumasa JN, Manzanillo PS, Peterson ND, Lundrigan T, Baughn AD, Cox JS, Keiler KC, Ribosome Rescue Inhibitors Kill Actively Growing and Nonreplicating Persister Mycobacterium tuberculosis Cells, ACS Infect Dis 3(9) (2017) 634-644.
[4] Shang Y, Cui D, Yi S, Opening tight junctions may be key to opening the blood-prostate barrier, Med Sci Monit 20 (2014) 2504-2507.
Questions for Authors
- Given that a blood-prostate barrier is a common consideration for drugs targeting STIs [4], were any assays done to characterise its impact on the pharmacokinetics on MBX-4132? Do you foresee that this will be an important issue to consider as MBX-4132 moves into further clinical development?
- In the in vitro tests for selectivity between gonorrhoeae and the various mammalian cell lines, how did the selectivity for mammalian cells compare to each other? Were any mammalian cell lines, other than HepG2 and HeLa, tested in vitro?
- What are the major routes of elimination for MBX-4132? Is it mostly metabolised or excreted? What are the key metabolites of MBX-4132? Does it mainly undergo Phase I or Phase II metabolism?
- Given the small molecular size and the good in vitro permeability of MBX-4132, do you think it will likely be able to penetrate the blood-brain barrier? Indeed, was this an important consideration in the design of this series of compounds?
doi: https://doi.org/10.1242/prelights.21990
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the microbiology category:
Circadian Clock Programming of Anticipatory Antiviral Immunity Gates Enteric Virus Infection Susceptibility
Owen Ang
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
Gut microbiome changes over the course of multiple sclerosis differentially influence autoimmune neuroinflammation
Carole Djagang et al.
Also in the pharmacology and toxicology category:
The Endocannabinoid System’s Contribution to Placebo Analgesia
Thomas Nicodemo Arrieta et al.
Small Molecule Agonists of TREM2 Reprogram Microglia and Protect Synapses in Human Alzheimer’s Models
Dina Kabbara
Snake venom metalloproteinases are predominantly responsible for the cytotoxic effects of certain African viper venoms
Daniel Osorno Valencia
preLists in the microbiology category:
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
BioMalPar XVI: Biology and Pathology of the Malaria Parasite
[under construction] Preprints presented at the (fully virtual) EMBL BioMalPar XVI, 17-18 May 2020 #emblmalaria
| List by | Dey Lab, Samantha Seah |
1
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the pharmacology and toxicology category:
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
COVID-19 / SARS-CoV-2 preprints
List of important preprints dealing with the ongoing coronavirus outbreak. See http://covidpreprints.com for additional resources and timeline, and https://connect.biorxiv.org/relate/content/181 for full list of bioRxiv and medRxiv preprints on this topic
| List by | Dey Lab, Zhang-He Goh |
1
Drug use in special populations
Any drugs that are being used in special populations: Patients with liver and kidney failure, in paediatrics, in geriatrics, and in pregnant or lactating patients. Includes the discovery of factors that could potentially affect drug use in these special populations.
| List by | Zhang-He Goh |
Toxicology of toxicants, existing therapeutics, and investigational drugs
Preprints that describe the toxicology of environmental pollutants and existing and upcoming drugs. Includes both toxicokinetics and toxicodynamics, as well as technological improvements that will help in the characterisation of this field.
| List by | Zhang-He Goh |
Anticancer agents: Discovery and clinical use
Preprints that describe the discovery of anticancer agents and their clinical use. Includes both small molecules and macromolecules like biologics.
| List by | Zhang-He Goh |
Advances in Drug Delivery
Advances in formulation technology or targeted delivery methods that describe or develop the distribution of small molecules or large macromolecules to specific parts of the body.
| List by | Zhang-He Goh |