








STAT3 promotes melanoma metastasis by CEBP-induced repression of the MITF pigmentation pathway
Posted on: 8 October 2018
Preprint posted on 20 September 2018
Article now published in Oncogene at http://dx.doi.org/10.1038/s41388-020-01584-6
Switching melanoma behaviour with STAT3: STAT3 represses the MITF pathway to drive metastasis
Selected by Hannah BrunsdonCategories: cancer biology
Introduction
Melanoma is the most aggressive form of skin cancer but despite advances in melanoma research, the prognosis for advanced melanoma remains poor, as tumours are highly resistant to chemotherapy. One reason for this is that melanomas can switch their phenotype between highly proliferative but non-invasive, to slowly proliferating and highly invasive, thus evading the actions of drugs and continuing to grow and metastasise[1].
A key genetic factor in this melanoma phenotype switching is the transcription factor MITF, which controls the development, differentiation and pigmentation of melanocytes, the cells from which melanomas occur. Melanomas with high MITF levels are characteristic of having the highly proliferative but non-invasive phenotype, whereas MITF-low melanomas proliferate slowly but are highly metastatic.
Therefore, understanding the regulation of MITF and its targets is important when designing therapies to halt melanoma growth and progression. A promising candidate for such a regulator is the transcription factor STAT3. 50% of melanoma patients have enhanced activation of STAT3, and in vitro, STAT3 activation causes cells to acquire melanoma-initiating and stem cell-like properties, thus promoting melanoma growth[2]. However, its precise role in vivo, and whether directly it regulates gene expression – particularly MITF – remains unclear.
Key findings
To investigate the effect of STAT3 loss in melanoma, the authors conditionally deleted the Stat3 gene in an existing genetic mouse model of human cutaneous melanoma to obtain the line Tyr::NRASQ61K;Ink4a-/- ; Stat3flox/flox; Tyr::Cre, which they termed Stat3Δ. Control mice were from the same genetic background, but lacked Tyr::Cre and so expressed Stat3 at normal levels.
Stat3Δ mice developed tumours significantly earlier than controls, and these tumours were reminiscent of MITF-high melanomas as they were more proliferative and pigmented, but non-invasive. Conversely, control mice developed more micrometastases in their lungs, phenotypically characteristic of MITF-low melanomas (see Figure below). Xenograft studies and in vitro invasion assays using Stat3Δ-derived cells supported these findings.
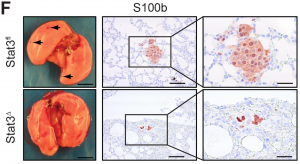
From preprint Figure 1F, with permission from the authors. Stat3 deletion reduces the formation of metastatic lung colonies (arrowed). Sections of 40 week old Stat3Δ mouse (termed Stat3fl here) lungs were stained with the melanoma marker S100b.
This led the authors to hypothesise that STAT3 activity might influence melanoma phenotype switching by regulating MITF expression. To test this, the authors compared the transcriptomes of cells isolated from Stat3Δ tumours to controls through analysis of a microarray dataset. Indeed, Stat3Δ cells had strongly upregulated levels of MITF and its targets, and in general, their transcriptome resembled MITF-driven proliferation signatures found in previous melanoma cohort studies.
Next, to explore the mechanism of this MITF upregulation in Stat3Δ cells, the authors performed ATAC-seq to see whether STAT3 or its downstream targets might directly bind to the Mitf promoter region to influence gene expression. Interestingly, an accessible binding region for the STAT3 targets CEBPA and CEBPB was found within the Mitf promoter region in control cells, but not in Stat3Δ cells. Moreover, rescuing Cebpa levels in Stat3Δ cells led to Mitf downregulation. This suggests that STAT3 deletion in melanoma causes epigenetic changes to chromatin, which leads to large changes in gene expression and tumour behaviour as a result of its targets CEBPA/B repressing the MITF pathway.
Finally, the authors showed that these findings have clinical relevance in human melanoma. shRNA-mediated knockdown of STAT3 in human melanoma cell lines recapitulated changes in gene expression seen in Stat3Δ cells. The authors also searched publicly available cancer genome databases to confirm STAT3-low/MITF-high expression signatures are found in humans. Interestingly, patients with poorer prognoses were more likely to have high levels of MITF, and low STAT3, CEBPA and CEBPB levels. This suggests that the balance between STAT3 and MITF in different stages of melanoma is important, and this could be a good target for designing new therapies.
Why I chose this preprint
Working in a Cancer Research institute, it is always disheartening to learn about how quickly melanoma patients develop resistance to treatment, and how aggressively their tumours can reoccur, despite huge efforts to search for chemo and immunotherapies to target and kill melanoma. From this preprint, and other studies on MITF in melanoma, it is worrying to learn that common therapies like BRAF inhibitors cause a reduction in MITF levels, and therefore could actually worsen outcomes if used in isolation. Therefore, I think this preprint highlights a potential target for regulating MITF levels in melanoma, but also shows there are huge challenges ahead in correctly balancing and timing drug treatment to prevent unwanted activation of genes which can exert large effects on tumour growth and survival.
Open questions and future directions
- The authors conclude that increased STAT3 activity drives metastasis through performing the ‘opposite’ experiments – deleting STAT3 and comparing it to wild type levels. I wonder if the authors have tried overexpressing STAT3 in a mouse mutant or cell line to show enhanced invasiveness compared to control and/or Stat3Δ tumours?
- I also wonder whether inducible deletion of STAT3, or the use of a STAT3 inhibitor in mice with established melanoma might cause a reversal of metastatic behaviour and shrink primary tumours? Or might it drive proliferation of the existing metastases and accelerate melanoma progression?
- This might be unanswerable yet, and so a bit of an unfair question, but how would one go about inhibiting the MITF and STAT3 pathways to treat melanoma? Would targeting one pathway after another be the best approach, or would it be better to target both pathways simultaneously to ‘cancel out’ their effects?
Further reading
- Kemper, K et al, 2014. Phenotype Switching: Tumor Cell Plasticity as a Resistance Mechanism and Target for Therapy. Cancer Research. 74(21)5937-5941
- Ohanna, M. et al., 2013. Secretome from senescent melanoma engages the STAT3 pathway to favour reprogramming of naïve melanoma towards a tumor-initiating cell phenotype. Oncotarget, 4(12)2212-2224
doi: https://doi.org/10.1242/prelights.5083
Read preprint (1 votes)
(1 votes) Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the cancer biology category:
Targeting CXADR-mediated AKT signaling suppresses tumorigenesis and enhances chemotherapy efficacy in Ewing sarcoma
TheLangeLab et al.
Dual inhibition of GTP-bound (ON) and GDP-bound (OFF) KRASG12C suppresses PI3Kα and leads to potent tumor inhibition
Luis Luna Ramírez
A Novel Chimeric Antigen Receptor (CAR) - Strategy to Target EGFRVIII-Mutated Glioblastoma Cells via Macrophages
Dina Kabbara
preLists in the cancer biology category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Anticancer agents: Discovery and clinical use
Preprints that describe the discovery of anticancer agents and their clinical use. Includes both small molecules and macromolecules like biologics.
| List by | Zhang-He Goh |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |