








Longitudinal single cell RNA-sequencing reveals evolution of micro- and macro-states in chronic myeloid leukemia
Posted on: 3 November 2025 , updated on: 4 November 2025
Preprint posted on 3 October 2025
A mathematical model translates aggregated single-cell RNA sequencing data into stages of disease
Selected by Charis QiCategories: bioinformatics, cancer biology, systems biology
Background
Single-cell RNA sequencing (scRNA-seq) has transformed the field of cancer research by allowing scientists to study gene expression of cancerous cells at the individual cell level [1][2][3]. However, identifying disease states is more challenging with scRNA-seq than with bulk RNA sequencing (RNA-seq). Bulk RNA-seq reveals different disease stages by providing a gene expression profile from a large population of cells [4]. In contrast, scRNA-seq data are derived from single cells and contain a lot of variability, making it much harder to identify disease stages [4].
In this preprint, Frankhouser and colleagues sought to understand how the single-cell transcriptome translates into the distinct disease states detected by bulk RNA-seq. Their approach was based on state-transition theory which suggests that while the individual cells exhibit variability, their disease states will become clear at the cell aggregate level. They focused on chronic myeloid leukemia (CML), a type of cancer that has two phases: it begins at the chronic phase (CP) and develops into the blast crisis (BC) phase [5]. Ultimately, they showed that scRNA-seq data can be used to reveal different disease states at the pseudobulk scale.
Key Highlights
Discrete CML stages are only visible at the cell aggregate level
Frankhouser and team first investigated CML with bulk RNA-seq using time-series gene expression data. Using Principal Component Analysis via Singular Value Decomposition (SVD), they identified that the biggest change in gene expression is associated with the shift from healthy to diseased states.
The researchers then examined a time-series scRNA-seq dataset, collected weekly from mice as they developed CP CML. Their initial goal was to map the disease’s progression from a healthy state to a diseased state through analyzing individual cells. However, their attempts showed that the patterns of change were due to differences in cell type rather than the stage of leukemia. When investigating the cells within each cell type, they were still unable to detect a unique state-transition between the healthy and diseased cells.
The researchers decided to combine the gene counts of all the single cells together into a pseudobulk sample to see if state-transition could be detected in aggregate. Using the SVD method, they were able to map the trajectory of the single-cell dataset from healthy to leukemic. They identified three distinct states within the trajectory: the Early state, the Transition state, and the Late state.
Each cell type contributes to CML state-transition
The authors investigated state-transition within each of the four cell types (B cells, T cells, myeloid cells, and stem cells) of the scRNA-seq data. Using SVD at the pseudobulk stage, they found that the trajectory states are clear when cells are aggregated together in each cell type. They then conducted a computer simulation to see how much each cell type influences the overall disease trajectory. They fixed one cell type at the healthy state while allowing the other cell types to naturally evolve. They compared the fixed simulation with the natural trajectory and saw that the simulated myeloid population caused the biggest information loss (a proxy for cell type importance), suggesting it plays the most important role in the CML state-transition. They also found that, although myeloid cells made the largest contribution, each cell type contributed to the overall leukemic state.
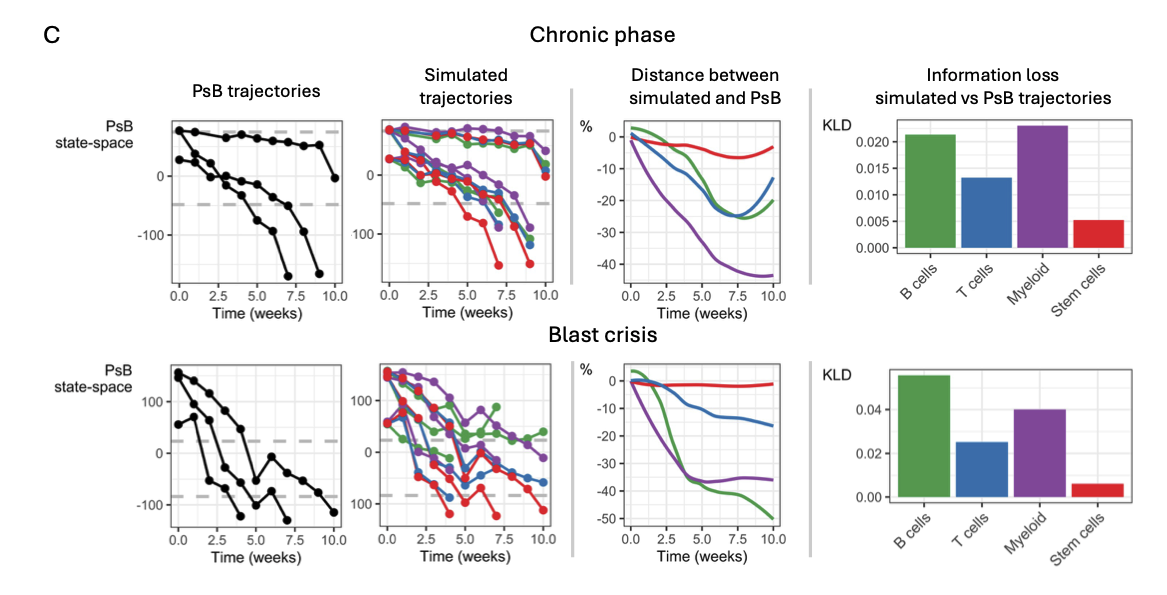
Figure 2C from the preprint – The computer simulation of the mouse CML model in the chronic phase (top) and blast crisis phase (bottom). The researchers used a principal component analysis (PCA) and time-ordering to fit a trajectory line through the pseudobulk (PsB) data points from healthy to sick. The graphs on the left show the simulated trajectory compared to the real trajectory, and the graphs on the right show the cell type contributions through information loss. Figure made available under a CC-BY-NC-ND 4.0 International license.
The researchers then confirmed these findings with a different CML mouse model that mimics the BC phase. Once again, they identified that state-transition can only be found at the cell aggregate level for single-cell data. They then separated the cells into their respective cell types and investigated the trajectory within each cell type. This time, when implementing the simulation, they found that the B cells, along with the myeloid cells, were the biggest drivers of state-transition.
State-transition approach is validated in human CML model
The authors decided to test their approach on human CML samples due to potential limitations of their mouse models. They analyzed scRNA-seq data from bone marrow stem and progenitor cells. They were unable to detect state-transitions when investigating the cells individually. However, when aggregated together, they were able to differentiate between the healthy state and the leukemic state.
The authors then adapted their computational simulation to work with the patient data. Using this simulation, they fixed each cell type to measure the information loss of the overall trajectory. They identified that common myeloid progenitors (CMP) and Pro B-cells contributed the most information to state-transition.
A mathematical model demonstrates cell-type contribution to state transitions
After demonstrating that the overall disease state is made up of each cell type’s contribution, the researchers built a mathematical model to formalize this relationship. The model is based on a concept of linear combination, where the state of the disease is the sum of the individual state of each cell type. For validation, the researchers implemented the model on the time-series CP CML mouse scRNA-seq dataset and found that the model perfectly recreated the previously measured disease trajectory.
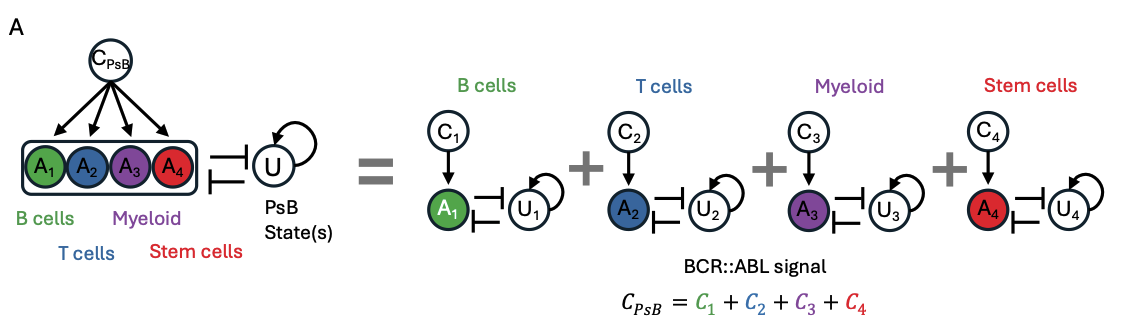
Figure 4A from the preprint – A visual representation of the mathematical model developed for pseudobulk state-transition. Figure made available under a CC-BY-NC-ND 4.0 International license.
Conclusion
By tracking CML progression with longitudinal single-cell RNA-seq, Frankhouser and team showed that the trajectory from healthy to diseased states can only be visible when single-cell microstates are aggregated into pseudobulk macrostates. By investigating the separate cell types of the cell aggregate, they found that each cell type contributes to state-transition. They built and validated a mathematical model representing the cell-type contribution. In the future, this model can pave the way for clinical applications, such as predicting disease development and treatment response.
Why this preprint is important
A notable strength of this paper is the authors’ ability to connect single-cell data directly to clinically relevant phenotypes. Typically, scRNA-seq data does not show a direct clinical relevance due to high variability at the individual cell level. However, when aggregated, the more relevant information of the disease was clearly shown. Additionally, as the authors mention, their framework showed that complex systems can be built from simple parts. Through their mathematical model they demonstrated that a complex, three-state disease trajectory can emerge from the combination of simple, single-state cells. The disease trajectory and systems-level approach discovered in this paper holds promise for future clinical applications, such as predicting a patient’s risk of a disease becoming more aggressive.
Questions for the authors:
- You define the macrostate by aggregating gene expression. Do you think other biological layers, like protein levels or cell-to-cell communication signals, are also part of this macrostate, and would incorporating them make the model even more accurate?
- How many single cells do you think are needed per sample to create a stable and reliable macrostate?
- In the computational simulation you conducted, is it possible that when you fix one cell type, the other cell types might over- or under-compensate for their absence? If so, how might these dynamic interactions affect the information loss you calculated for each cell type?
References:
[1] Li, L., Xiong, F., Wang, Y., Zhang, S., Gong, Z., Li, X., He, Y., Shi, L., Wang, F., Liao, Q., Xiang, B., Zhou, M., Li, X., Li, Y., Li, G., Zeng, Z., Xiong, W., & Guo, C. (2021). What are the applications of single-cell RNA sequencing in cancer research: a systematic review. Journal of experimental & clinical cancer research : CR, 40(1), 163. https://doi.org/10.1186/s13046-021-01955-1
[2] Zhang, Y., Wang, D., Peng, M., Tang, L., Ouyang, J., Xiong, F., Guo, C., Tang, Y., Zhou, Y., Liao, Q., Wu, X., Wang, H., Yu, J., Li, Y., Li, X., Li, G., Zeng, Z., Tan, Y., & Xiong, W. (2021). Single-cell RNA sequencing in cancer research. Journal of experimental & clinical cancer research : CR, 40(1), 81. https://doi.org/10.1186/s13046-021-01874-1
[3] Chang, X., Zheng, Y., & Xu, K. (2024). Single-Cell RNA Sequencing: Technological Progress and Biomedical Application in Cancer Research. Molecular biotechnology, 66(7), 1497–1519. https://doi.org/10.1007/s12033-023-00777-0
[4] Tzec‐Interián, J. A., González‐Padilla, D., & Góngora‐Castillo, E. B. (2025). Bioinformatics perspectives on transcriptomics: A comprehensive review of bulk and single‐cell RNA sequencing analyses. Quantitative Biology, e78. https://doi.org/10.1002/qub2.78
[5] Michor F. (2007). Chronic myeloid leukemia blast crisis arises from progenitors. Stem cells (Dayton, Ohio), 25(5), 1114–1118. https://doi.org/10.1634/stemcells.2006-0638
doi: https://doi.org/10.1242/prelights.41929
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
Temporal degradation of PRC2 uncovers specific developmental dependencies
María Mariner-Faulí
Science should be machine-readable
Theodora Stougiannou
Remote homology and functional genetics unmask deeply preserved Scm3/HJURP orthologs in metazoans
Reinier Prosee
Also in the cancer biology category:
Dual inhibition of GTP-bound (ON) and GDP-bound (OFF) KRASG12C suppresses PI3Kα and leads to potent tumor inhibition
Luis Luna Ramírez
A Novel Chimeric Antigen Receptor (CAR) - Strategy to Target EGFRVIII-Mutated Glioblastoma Cells via Macrophages
Dina Kabbara
Taxane-Induced Conformational Changes in the Microtubule Lattice Activate GEF-H1-Dependent RhoA Signaling
Vibha SINGH
Also in the systems biology category:
Human single-cell atlas analysis reveals heterogeneous endothelial signaling
Charis Qi
Longitudinal single cell RNA-sequencing reveals evolution of micro- and macro-states in chronic myeloid leukemia
Charis Qi
Environmental and Maternal Imprints on Infant Gut Metabolic Programming
Siddharth Singh
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the cancer biology category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Anticancer agents: Discovery and clinical use
Preprints that describe the discovery of anticancer agents and their clinical use. Includes both small molecules and macromolecules like biologics.
| List by | Zhang-He Goh |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |
Also in the systems biology category:
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Pattern formation during development
The aim of this preList is to integrate results about the mechanisms that govern patterning during development, from genes implicated in the processes to theoritical models of pattern formation in nature.
| List by | Alexa Sadier |