








PHYTOMap: Multiplexed single-cell 3D spatial gene expression analysis in plant tissue
Posted on: 2 September 2022 , updated on: 5 September 2022
Preprint posted on 30 July 2022
Low cost – high resolution: 3D single-cell mapping of up to 100 gene targets in an intact Arabidopsis root with PHYTOMap
Selected by Gwendolyn K. Kirschner, Marc SomssichCategories: bioengineering, molecular biology, plant biology
Background:
To ultimately understand the function of genes in a tissue and organ context, it is crucial to not only quantify gene expression in single cells, but also to identify the individual positions of these cells within the intact tissue. Isolation of specific cells for pooled RNA sequencing (RNAseq) in plants started in the early 2000s, for instance by using laser-capture microdissection to cut and isolate the tissue of interest with a laser beam under direct microscopic visualization, capturing subsets of cells via fluorescence-activated cell sorting (FACS), or isolating individually tagged nuclei (INTACT) (Asano et al., 2002; Birnbaum et al., 2003; Deal and Henikoff, 2011). The subsequently developed single-cell RNAseq (scRNAseq) technique was first applied to mouse primordial germ cells in 2009, and adapted for the use in plants a couple of years later, using isolated protoplasts which were manually collected based on a fluorescent reporter under a microscope (Tang et al., 2009; Brennecke et al., 2013; Efroni et al., 2015).
scRNAseq allows for the classification of cells into populations with similar transcriptomic profiles, thereby enabling the identification of certain cell types, such as the epidermis. Since the sampled cells are removed from their tissue context, however, all positional information for these populations is lost. This makes it challenging to classify and analyse cell populations that do not contain certain marker genes, for instance in plants that are less well-studied than the common model plant Arabidopsis. Furthermore, if experimental conditions only affect a subset of cells within a population, e.g. by a certain treatment, it is important to know where these affected cells are located, and if they form a spatially confined cluster within the population and tissue.
To accurately interpret scRNAseq data, the observed transcriptomic responses of individual cells need to be mapped back to the intact tissue. To date, the most common technique to spatially visualize gene expression in planta is the use of transgenic lines expressing transcriptional reporter proteins for the genes of interest. However, the creation of transgenic reporter lines is time consuming and tedious, and for some plant species even impossible. Another classic approach applicable to all plant species is RNA in situ hybridization, in which the mRNA of interest is directly targeted by RNA probes that can be detected by antibodies. However, mapping the transcription of only a single gene is insufficient to describe a cell population as observed in an scRNAseq experiment. Spatial transcriptomics, either by using spatially barcoded arrays or single-molecule fluorescence in situ hybridisation, are challenging because they require a single cell layer or thin sections, which is laborious in plant tissues and may again insert artifacts due to the sectioning. Additionally, with these techniques, information of the tissue environment is lost.
Ideally, spatial gene expression analysis of a large number of genes would be performed simultaneously at a single-cell resolution in an intact tissue context with minimal labor and financial requirements. And while this may sound illusory, in the publication highlighted here, Nobori et al. attempt to do just that with their PHYTOMapnyTM*, a method to spatially map the expression of multiple genes in whole-mount plant tissue at a single-cell level without the need for creating transgenic plant lines (Nobori et al., 2022). They develop a protocol for the Arabidopsis root tip and validate their method by mapping the gene expression of established marker genes and collating the expression to an scRNAseq dataset.
How a PHYTOMapnyTM is produced:
To produce a PHYTOMapnyTM, the root tissue is fixed and permeabilized, as it is for whole-mount RNA in situ hybridisation experiments. Hybridisation is performed with Specific Nucleic AcId Ligation (SNAIL) DNA probes targeted at the mRNAs of interest, which are labeled with gene-specific barcodes (Figure 1A). In the next step, the SNAIL probes are circularized and amplified in situ, incorporating amine-modified nucleotides into the amplicons to stably cross-link them to the cellular protein matrix. To then localize these barcoded amplicons, a multi-round sequencing-by-hybridization method is used, for which in every round four bridge probes are hybridized to four respective DNA barcodes, and then each bridge probe is tagged with one of four different fluorescent probes (Figure 1B). In an imaging step, using a standard confocal microscope, the four different targets can thus be detected in four different channels via their respective fluorescent probes. After the imaging, the probes are stripped and the next batch of bridge and fluorescent probes, targeting the next four DNA barcodes, are hybridized for the next round of imaging (Figure 1C). And so, with each round of probing and imaging, more transcripts can be added to the detailed PHYTOMapnyTM of one intact root.
The authors validated their approach by detecting transcripts of genes with well-known expression patterns, such as markers for different tissue types, and from a published scRNAseq dataset (Shahan et al., 2022). Additionally, they explored the multiplexing capacity of the method by targeting 28 genes in 7 rounds of hybridisation/imaging/stripping. For the combined analysis of the images from all rounds in one whole-mount image, the team developed an automated computational pipeline. For this, all images were registered in 3D, based on cell wall staining information cell segmentation was performed, and detected spots for the different transcripts were assigned to individual cells. According to the authors, the comparison between five roots yielded highly robust and reproducible data, detecting comparable numbers of transcripts for each RNA species between the different biological samples (See Movie 1). The data could be visualized as a Uniform Manifold Approximation and Projection (UMAP) without the input of spatial information and the clusters represent major cell types and developmental stages.
Why this preprint is important:
PHYTOMapnyTM will allow researchers to spatially and reproducibly map the expression of multiple genes at single-cell resolution within the same biological sample – an essential tool to get the most out of scRNAseq data. In this way, gene clusters that are identified in transcriptomic experiments can be assigned to specific tissues and cell groups in the intact plant. Furthermore, for Arabidopsis, the whole workflow can be performed at minimal time and labor costs. Time at the bench for the experimental work is similar to established RNA in situ hybridisation methods (Stahl and Simon, 2010): 4-5 days for sample preparation; for imaging, regular confocal microscopes can be used, with every round of imaging taking around 3 hours per root tip or 5 hours per five root tips. Using similar methods, it was shown that more than 25 rounds with new bridge probes and fluorescent probes are possible, so PHYTOMapnyTM can possibly map up to 25 x 4 =100 genes (Lee et al., 2015). As for the cost, commercially available tools to map the expression of several genes to tissues is offered by several companies, e.g. by localizing gene expression using 100-plex combinatorial single molecule fluorescence in situ hybridization. However, the cost for such commercial offers usually tallies up to several thousand dollars, and they are thus not affordable for many research groups. In contrast, for a 28 gene PHYTOMapnyTM, the authors calculate the cost to an approximate total of $80, and $230 for 96 genes, making this an experiment affordable for a broad range of researchers. Furthermore, by using intact plants, any artifacts or information loss due to sectioning of the tissue can be eliminated.
Challenges and future directions:
One major limit of PHYTOMapnyTM at this stage is that it has so far only been tested on Arabidopsis root tips. A main challenge therefore lies in the adaptation of the technique to other tissues, and, even more importantly, other plant species, especially non-model plants. The in vivo analysis and validation of gene expression using (e.g.) fluorescent reporter lines is routinely done in Arabidopsis (albeit simultaneously for 1-4 genes, not 1-100), but poses a significant challenge to researchers working on more complex plants that cannot (easily) be transformed. PHYTOMapnyTM holds the potential to massively accelerate research on such plants, if it can indeed be adapted to them. However, it is not clear if SNAIL probe and dye penetration would work as well in other tissue than the relatively small and thin root tip, which furthermore has the advantage of incomplete endodermal barrier formation, facilitating uptake of probes and dyes into the vasculature. More complex roots may require additional sectioning steps. For most plant species, RNA in situ hybridisation is still based on microtome tissue sectioning after embedding in paraffin (Zöllner et al., 2021), which results in flat layers of a few cells and therefore the 3D resolution of the approach would be lost. For these sections, the PHYTOMapnyTM protocol must presumably be highly modified. Another approach to deal with thick tissues is to section them by vibratome after the fixation without paraffin embedding, which has been used for whole-mount RNA in situ hybridisation on date palm tissues (Xiao et al., 2019). For this, only slight changes in the PHYTOMapnyTM protocol might be sufficient, and the 3D resolution would partially be maintained, because the sections consist of multiple cell layers.
First author Tatsuya Nobori has also added a thread on Twitter to introduce PHYTOMapnyTM: https://twitter.com/nobolly/status/1553408840237846528
In summary, spatial transcriptomics in plants is still in its infancy, but the PHYTOMapnyTM presented by Nobori et al. is a big step in the right direction.
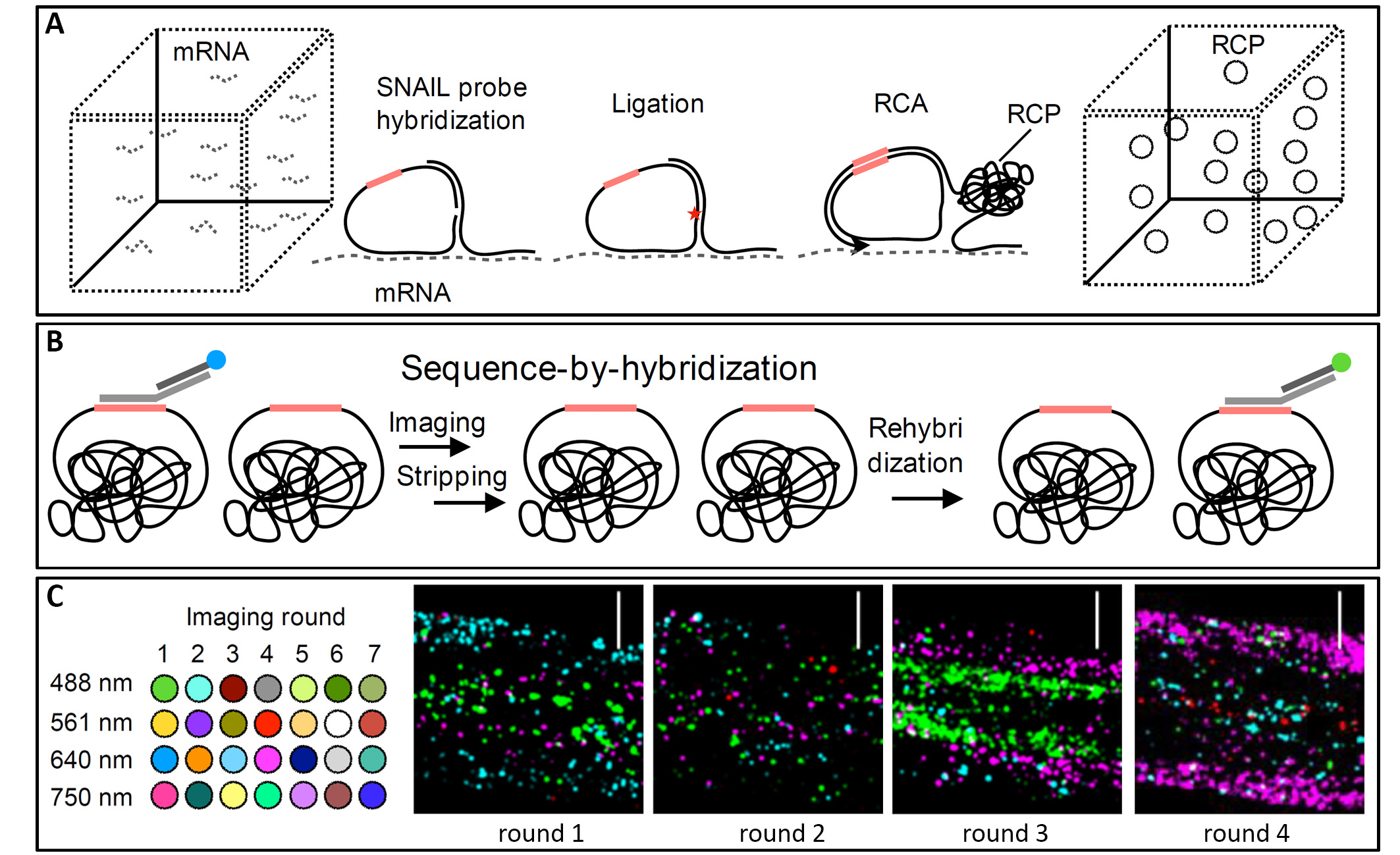
Figure 1: PHYTOMapnyTM concept and imaging results.
A) Experimental approach of PHYTOMapnyTM for probe hybridisation, ligation and amplification in situ. B) Sequence-by-hybridisation chemistry for sequential rounds of imaging by probing, imaging, and stripping. C) Set-up of the imaging rounds (left) and representative images of Arabidopsis root tips at different imaging rounds, with the four different fluorescent probes in blue, pink, green and red, shown as maximum exposure of z-planes at the same position (right), scale bars 30 μ Modified from (Nobori et al., 2022).
Movie 1: Proof of concept by mapping of four different genes per root; displayed are 3D projections of the root tip. Taken from (Nobori et al., 2022).
* = not yet TradeMarked
References:
Asano, T., Masumura, T., Kusano, H., Kikuchi, S., Kurita, A., Shimada, H. and Kadowaki, K.I. (2002) Construction of a specialized cDNA library from plant cells isolated by laser capture microdissection: Toward comprehensive analysis of the genes expressed in the rice phloem. Plant J., 32, 401–408.
Birnbaum, K., Shasha, D.E., Wang, J.Y., Jung, J.W., Lambert, G.M., Galbraith, D.W. and Benfey, P.N. (2003) A Gene Expression Map of the Arabidopsis Root. Science (80-. )., 302, 1956–1960.
Brennecke, P., Anders, S., Kim, J.K., et al. (2013) Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods, 10, 1093–1098.
Deal, R.B. and Henikoff, S. (2011) The INTACT method for cell typeg-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat. Protoc., 6, 56–68.
Efroni, I., Ip, P.L., Nawy, T., Mello, A. and Birnbaum, K.D. (2015) Quantification of cell identity from single-cell gene expression profiles. Genome Biol., 16, 1–12.
Lee, J.H., Daugharthy, E.R., Scheiman, J., et al. (2015) Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc., 10, 442–458.
Nobori, T., Oliva, M., Lister, R. and Ecker, J.R. (2022) PHYTOMap : Multiplexed single-cell 3D spatial gene expression analysis in plant tissue. bioxriv, 1–25.
Shahan, R., Hsu, C.W., Nolan, T.M., et al. (2022) A single-cell Arabidopsis root atlas reveals developmental trajectories in wild-type and cell identity mutants. Dev. Cell, 57, 543-560.e9.
Stahl, Y. and Simon, R. (2010) mRNA detection by whole mount in situ hybridization (WISH) or sectioned tissue in situ hybridization (SISH) in Arabidopsis. Methods Mol. Biol., 655, 239–251.
Tang, F., Barbacioru, C., Wang, Y., et al. (2009) mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods, 6, 377–382.
Xiao, T.T., Raygoza, A.A., Pérez, J.C., et al. (2019) Emergent protective organogenesis in date palms: A morpho-devo-dynamic adaptive strategy during early development. Plant Cell, 31, 1751–1766.
Zöllner, N.R., Bezrutczyk, M., Laureyns, R., Nelissen, H., Simon, R. and Frommer, W.B. (2021) An RNA in situ hybridization protocol optimized for monocot tissue. STAR Protoc., 2.
doi: https://doi.org/10.1242/prelights.32621
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioengineering category:
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Detergent-Triggered Membrane Remodelling Monitored via Intramembrane Fluorescence De-Quenching
Cyntia Alves Conceição, Marcus Oliveira
A Novel Chimeric Antigen Receptor (CAR) - Strategy to Target EGFRVIII-Mutated Glioblastoma Cells via Macrophages
Dina Kabbara
Also in the molecular biology category:
BAF complexes maintain accessibility at stimulus-responsive chromatin and are required for transcriptional stimulus responses
Dina Kabbara
Aging increases ovarian cancer growth, metastasis, and immunosuppression that can be alleviated by inhibiting hedgehog signaling
Zoha Sadaqat
A pre-rRNA positive feedback loop drives malignant ribosome biogenesis
Vaishali Grewal
Also in the plant biology category:
Structure of chlorophyll synthase in complex with the LHC-like protein HliD
Orestis Savva
A drought stress-induced MYB transcription factor regulates pavement cell shape in leaves of European aspen (Populus tremula)
Jeny Jose
Actin Counters Geometry to Guide Plant Cell Division
Jeny Jose
preLists in the bioengineering category:
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
3D Gastruloids
A curated list of preprints related to Gastruloids (in vitro models of early development obtained by 3D aggregation of embryonic cells). Updated until July 2021.
| List by | Paul Gerald L. Sanchez and Stefano Vianello |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Advances in microscopy
This preList highlights exciting unpublished preprint articles describing advances in microscopy with a focus on light-sheet microscopy.
| List by | Stephan Daetwyler |
Also in the molecular biology category:
Developmental regulation: molecular and ecological niches
This conference was held at the Station Biologique de Roscoff (France) and brought together researchers exploring how diverse niche environments shape developmental processes across scales. Spanning topics from ecological and metabolic influences to signalling networks, mechanics and gene regulation, the meeting highlighted the interplay between intrinsic and extrinsic factors in controlling cell fate and tissue organisation. This preList gathers preprints discussed by speakers and poster presenters during the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |
Also in the plant biology category:
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
The Society for Developmental Biology 82nd Annual Meeting
This preList is made up of the preprints discussed during the Society for Developmental Biology 82nd Annual Meeting that took place in Chicago in July 2023.
| List by | Joyce Yu, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
SDB 78th Annual Meeting 2019
A curation of the preprints presented at the SDB meeting in Boston, July 26-30 2019. The preList will be updated throughout the duration of the meeting.
| List by | Alex Eve |