








Precise genomic deletions using paired prime editing
Posted on: 27 January 2021
Preprint posted on 2 January 2021
Article now published in Nature Biotechnology at http://dx.doi.org/10.1038/s41587-021-01025-z
PRIME-Del: Two “pegs” are better than one – precise genome deletions with paired prime editing guide RNA
Selected by Fabio LiberanteCategories: genomics, molecular biology, synthetic biology
Context
Genome engineering has seen an explosion in recent years thanks to the Nobel prize-winning discovery of CRISPR/Cas proteins. Their ease of use has enabled a myriad of new functional studies exploring genes, domains, enhancers and structural elements of DNA sequence. The original programmable DNA-binding technology has spawned numerous novel molecular tools, including activators, inhibitors, silencers, repressors, interaction-creators and base editors. At the end of 2019 a new CRISPR tool took centre-stage, prime editing. This ingenious use of a Cas-reverse transcriptase fusion and an extended guide RNA (pegRNA) enabled very simple substitutions, insertions and deletions at defined loci, no longer relying on DNA donors or limited by the one-at-a-time transitions or transversions of base editors. Another advantage is the fact that the site to be targeted and the template are encoded in a single pegRNA. The paper has already received over 200 citations in just over 12 months showing its usefulness. Despite its potential, it’s still unable to delete large regions efficiently. Similarly, the use of paired guides with typical CRISPR/Cas to trigger simultaneous double-strand breaks is fraught with its own problems. The authors of this preprint present an application of prime editing that enables large precise deletions and simultaneous insertions along with a tool for designing the necessary pegRNAs.
Key ideas
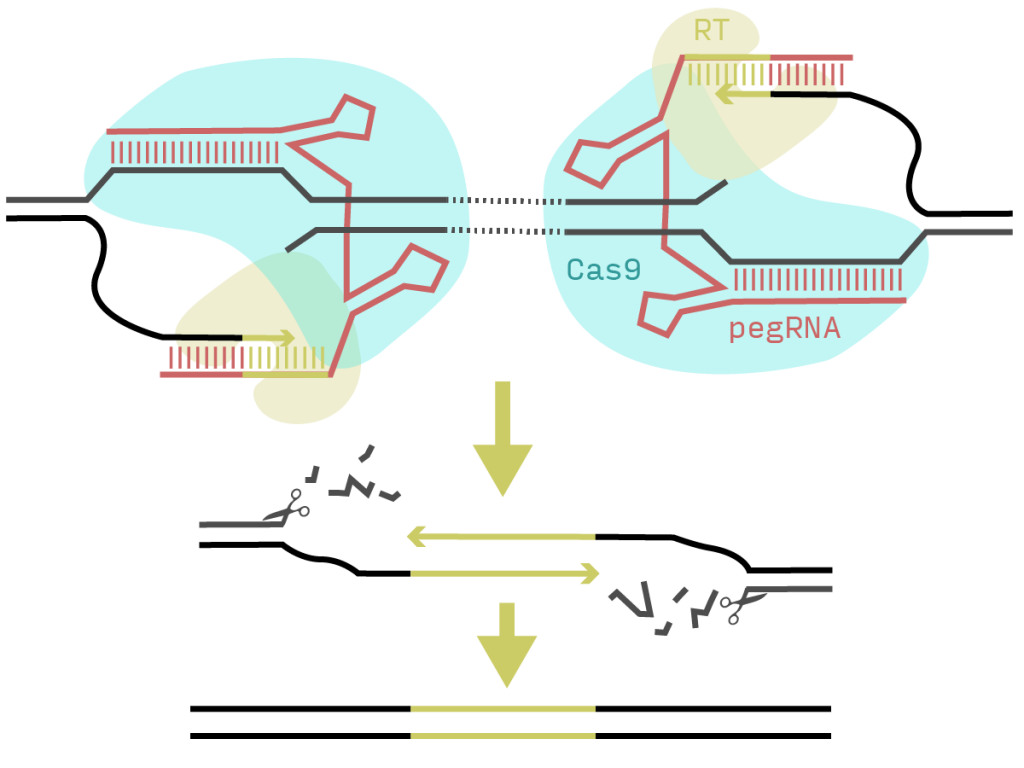
PRIME-Del – creating deletions with prime editing and pairs of pegRNAs
Prime editing uses a Cas9 nickase and reverse transcriptase (RT) fusion (PE2) together with an extended guide RNA (pegRNA). The Cas9 nicks the DNA and the RT extends the free end using the 3’ region of the pegRNA as primer and template. The authors of this preprint realised that they could use a second pegRNA on the opposite strand at an arbitrary distance and insert a matching overhang that should trigger a base-pair-accurate deletion of the intervening region (Figure 1).
To test this approach they assembled a dual pegRNA vector using the U6 and H1 RNA PolIII promoters on the same plasmid. Initially, they targeted the eGFP cassette present on the prime editor (PE2) expressing vector. The PRIME-Del method proved successful and showed an efficiency ranging between 38% and 77% for 24 to 546bp deletions for this episomal target. Interestingly, larger deletions showed a higher efficiency.
Once the authors had shown the system worked they attempted to use it to modify a genomic target and so integrated a similar eGFP cassette using lentivirus. While successful, this was unfortunately much less efficient at only 10%. However, targeting the HPRT1 native genomic locus yielded much better deletion rates of up to 25%. This likely illustrates the importance of the target and its genomic context.
PRIME-Del is more precise than CRISPR/Cas9
The authors next compared PRIME-Del to previous techniques, which use a pair of regular guide RNAs and double-strand cleaving Cas9. This showed that Cas9 yielded ~15% indels even without successful deletion of the target region vs only 2.2% for PRIME-Del. The overall error rate for both was higher if the region was deleted, but still nearly four times lower for PRIME-Del. While the much greater precision of the PRIME-Del approach is impressive, most use cases for cells modified in such a way requires sub-cloning to ensure that only cells with the exact mutation/deletion are studied. When the efficiencies for both systems are looked at purely in terms of the overall percentage of cells with the exact deletion they are not dissimilar. In the example presented for the 252 bp deletion in HPRT1 the percentage of perfect deletions are approximately 25% vs. 19% for PRIME-Del and Cas9, respectively. So while PRIME-Del is very precise, it is only marginally more efficient than Cas9, despite its more complex design.
I suspect the authors were disappointed with the small increase in efficiency initially offered by PRIME-Del. Therefore they attempted to boost it by constitutive expression of both the prime editor and the dual pegRNAs. This is made viable by the high precision of the system as random “dead-end” mutations don’t accumulate quickly. Promisingly, this proved a success and yielded correct 118 bp deletions in nearly 50% of K-562 cells after 19 days in culture.
Benefits of PRIME-Del
Despite the limited overall efficiency in transient PRIME-Del, there are certain circumstances where greater precision is indeed much more important. One obvious example is in any future applications in gene therapy – where any unintended mutations could be dangerous. While the authors do not directly recommend PRIME-Del for use in gene therapy, they demonstrate its potential by deleting a CGG-repeat expansion region in the FMR1 gene implicated in Fragile-X syndrome. A second example where precision is important is in any multiplex studies where single cell cloning is not possible – for example if targeting various protein domains for deletion, reducing the number of spurious indels or knockouts simplifies downstream analysis. Further, PRIME-Del, and prime editing in general, are likely better suited to high-throughput screening as they only induce single-strand DNA nicks, which is often less toxic to cells, a common limiting factor to using regular CRISPR/Cas in certain cell types.
Perhaps one of the most exciting innovations of the PRIME-Del system is that it allows simultaneous insertion of novel sequence at the point of deletion. The authors demonstrated successful insertion of up 30bp using this technique. It doesn’t rely on a separate repair template and does not require homologous repair to incorporate foreign DNA. Encouragingly, the authors show that if the deletion is successful then so is the insertion in over 99% of cases.
Challenges and limitations
In terms of pitfalls, the authors identify some designs whereby PRIME-Del can trigger a relatively high frequency of small duplications, mainly due to high GC content at the 3’ ends of the engineered overlaps. This is something that their design tool seeks to minimise.
While the design of a pegRNA is more complicated than a regular guide RNA and a paired pegRNA PRIME-Del design even more so, the authors have helpfully written a Python-based tool [https://github.com/shendurelab/Prime-del] (and web instance https://primedel.uc.r.appspot.com/]) which simplifies the process.
The authors do highlight some limitations with their system. Primarily, that paired pegRNAs require the presence of PAM sites near each end of the deletion and on opposing strands. They suggest that this might eventually be solved using a “PAM-less” prime editor.
What I like about this work
One of the great things about this work is the simplicity of the idea, which is then borne out in a series of well thought-out experiments. Authors addressed a typical concern about PCR bias in quantifying deletion efficiency by using a clever linear amplification step to label molecules with a 15 bp UMI – it turned out there were only modest effects anyway! PRIME-Del is tested in the proper range of contexts – episomal, integrated synthetic construct and native genomic loci. Finally, the preprint comes with an accompanying software tool to enable others to quickly design and test their own PRIME-Del pegRNAs.
Open questions
- The largest deletion demonstrated was 710 bp. What are the upper size limits for PRIME-Del?
- Simultaneous insertion at the point of deletion is useful, but what is the largest insert?
- The authors mention that PAM sites need to be present near deletion junctions, but how far away can the sites be? Especially important in AT-rich genomes.
- Did the authors consider trying a multiple transfection (or sustained expression) approach with regular Cas9 to demonstrate the “dead ends” that should accumulate? Also, did they compare PRIME-Del to a single pegRNA for deletions?
- The authors used H1 and U6 promoters to express the two pegRNAs. Did they try other PolIII promoters, such as mU6 or 7SK, or even Csy4 arrays?
- Interestingly, it seems that in most cases larger deletions were more efficient. Do the authors have any suspicions as to why? Is it simple steric hindrance?
- The overall efficiency seems to vary widely depending on cell type and genomic context. Did the authors investigate what factors might contribute to this variation? Guide RNA score, chromatin structure etc.?
doi: https://doi.org/10.1242/prelights.27128
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the genomics category:
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
Also in the molecular biology category:
Disordered protein COSA-2 maintains crossover-specific repair compartments to ensure meiotic crossover maturation
Chee Kiang Ewe
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Defective BRCA1-mediated DNA end resection drives tandem duplication formation and FANCM synthetic lethality
Marta San Martin
Also in the synthetic biology category:
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Enzymatic bromination of native peptides for late-stage structural diversification via Suzuki-Miyaura coupling
Zhang-He Goh
Enhancer cooperativity can compensate for loss of activity over large genomic distances
Milan Antonovic
preLists in the genomics category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |
Also in the molecular biology category:
Developmental regulation: molecular and ecological niches
This conference was held at the Station Biologique de Roscoff (France) and brought together researchers exploring how diverse niche environments shape developmental processes across scales. Spanning topics from ecological and metabolic influences to signalling networks, mechanics and gene regulation, the meeting highlighted the interplay between intrinsic and extrinsic factors in controlling cell fate and tissue organisation. This preList gathers preprints discussed by speakers and poster presenters during the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |
Also in the synthetic biology category:
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Advances in Drug Delivery
Advances in formulation technology or targeted delivery methods that describe or develop the distribution of small molecules or large macromolecules to specific parts of the body.
| List by | Zhang-He Goh |