








Rapid discovery of synthetic DNA sequences to rewrite endogenous T cell circuits
Posted on: 18 June 2019 , updated on: 19 June 2019
Preprint posted on 12 April 2019
T-cells for everyone! A platform to screen functionality of novel DNA sequences in T-cells.
Selected by Pavithran RavindranCategories: bioengineering, cancer biology
Background
The engineering of a patient’s immune cells to specifically target and kill cancer cells has become a field of research with a lot of momentum in recent years. Ever since the advent of genetically engineering a patient’s T cells to produce chimeric antigen receptors (CAR-T cells), which have even shown efficacy in clinical, a number of researchers have developed various synthetic receptors that extend this initial breakthrough1,2. For instance, one group developed a receptor that uses the immunosuppressive signal, TGFβ, and rewires the signal into an activating signal for T-cells3. Furthermore, a lot of beautiful work has been done to develop novel receptors that can be used to target specific combinations of cancer targets to reduce off-target effects4–6. Although this research has exploded in the past few years, it is clear that developing and validating novel DNA sequences for receptors is quite difficult and new methods to do so would help this field tremendously. The authors of this preprint attempt to do just this. They first develop a method to massively parallelize the insertion of novel DNA sequences into the genome of T-cells. They then use this to create a library of T-cells with novel receptors and put this library through a number of selection pressures to obtain known and novel DNA insert sequences. Finally, they even go as far as to take this library and use it in vivo with a tumor mouse model to identify possibly new mechanisms of helping T-cells navigate the tumor microenvironment.
Key Findings
In this preprint, the authors set out to develop a method to rapidly develop novel DNA sequences in T-cells that encode proteins to increase T-cell functionalities and allow them to combat the ever-evolving landscape of cancer. The authors had recently published a method that allows them to edit the genomes of T-cells using CRISPR-Cas9 technology with remarkable efficiency7. As a first step toward the goal of creating a screening platform for specific T-cell functions, the authors expanded their previous work to understand the breadth of genes that could be screened. They utilized an 800 base-pair reporter gene and CRISPR tagged this cassette to 91 genes and found that 88% of the loci were amenable to genomic integration of the reporter. Furthermore, they were able to analyze the efficiency for each gene and found that gRNA sequence, RNA expression of the target gene and chromatin accessibility were able to explain the variation. Next, they wanted to test the depth to which they could study novel sequences to reprogram T-cells. Specifically, they wanted to create targeted knock-in that integrate synthetic elements with existing sequences, Genetically Engineered Endogenous Proteins (GEEPs), by varying the gene regulation, gene product, or the intra/extracellular domains of endogenous T-cell receptors. As a proof of principle, the authors decided to engineer the IL2RA gene and, remarkably, they were able to increase expression of the receptor by knocking-in a constitutive promoter, and impart the stimulus dependent expression of the endogenous gene to a novel receptor by placing the new receptor in front of the endogenous protein with a self-cleaving peptide tag. Furthermore, they were able to completely replace entire domains of the gene. Overall, the authors undertook and accomplished the herculean task of CRISPR tagging 91 genes and then developing GEEPs with IL2RA.
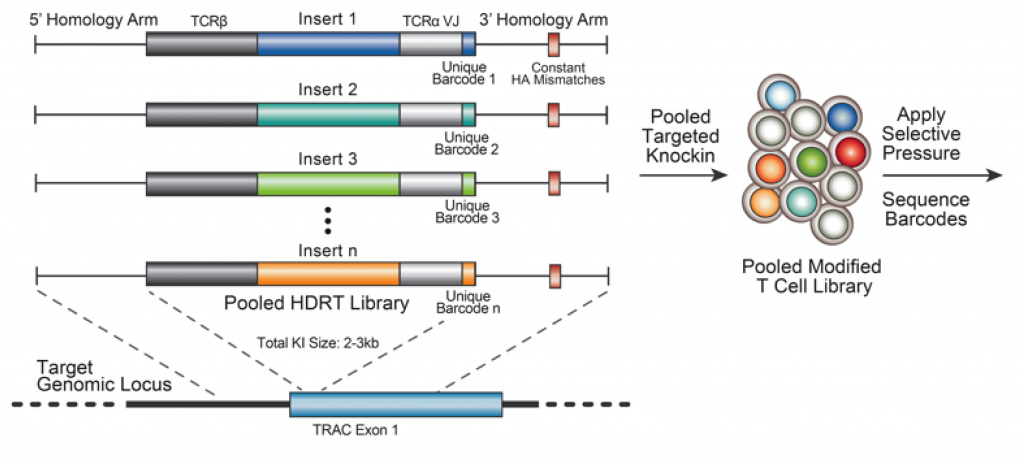
With these results, the authors were situated to develop a screen in which they could screen through various polycistronic DNA sequences that modify T cell function for particular therapeutic contexts (Figure 1). As their test bed, the authors decided to modify the endogenous TRAC gene with new TCRβ and TCRα sequences alone with a novel receptor inserted in the middle along with a barcode sequence. This was then incorporated into T-cells and the library was tested for knock-in of the various inserts. To accomplish this, the authors introduced a small sequence of DNA in the homology arms that does not match the endogenous sequence far from the insert location. Based on the fact that with on-target HDR, integration does not incorporate the entire homology arm while template switching does, the authors could sequence this small sequence of DNA and if they got back the original sequence, then on-target HDR occurred, while if the new sequence was there, there may have been off-target integration. The authors knocked in 36 different inserts into T-cells containing dominant negative receptors, synthetic receptors, transcription factors and metabolic regulators. With this pooled library of T-cells, the authors then wanted to ask what DNA sequences would become enriched after anti-CD3/CD28 stimulation. The authors found a novel Fas “switch” receptor with a 4-1BB intracellular domain. This switch receptor takes in the input of FASL, which activates apoptosis in T-cells upon binding to Fas, and redirects it into a T-cell persistence output. This result was highly reproducible and highlights the versatility of this screening platform to identify novel knock-in synthetic receptors that promote specific phenotypes in particular environments.
The authors took this one more step further by applying their screen in an in vivo context. The authors introduced their T-cell library into a mouse bearing human melanoma expressing the NY-ESO-1 antigen. Upon extraction of T-cells from the tumor 5 days after in vivo tumor expansion, the authors identified a unique metabolic protein, MCT4, that had not shown enrichment in any of their other screens. They also found 3 strong hits (an anti-suppressive TGF R2-41BB chimera, an anti- apoptotic FAS-41BB chimera, and the transcriptional program altering TCF7) that they decided to do individual validation of. Remarkably, each were not only able to increase T-cells’ ability to grow but also to eliminate the melanoma cancer cells. Overall, this work enables the use of endogenous genome editing of T-cells to find novel DNA sequences that provide synthetic T-cells an advantage to kill cancer cells in a context dependent manner.
Why I chose this preprint
I chose this preprint simple due to the herculean effort that this work seem to encompass. Not only did the authors of this preprint make a protocol that allowed them to CRISPR tag almost 90 genes in T-cells, they also developed a way to make large insertions into T-cells, created a library of T-cells with endogenous and novel receptors, and screened the library both in in vitro and in vivo contexts. The paper was very easy to read and the figures clearly communicated the science being showcased. Overall, the authors managed to take very complex and massive amount of data and communicate it in a very digestible manner; thus, this paper was truly a joy to read.
Questions for the authors
- What is the biggest insert that you were able to do with your protocol and do you think this is the limit? Could one do not just a single novel insert but multiplex to understand the combinatorial effects of different receptors/TFs/metabolic regulators etc.? Better yet, could one screen for combinatorial gate logic (AND, ANDNOT, etc.) using this?
- How easily does your protocol transfer to different types of cells? Could you use this CRISPR protocol to modify multiple types of immune cells to understand their effects in combination with one another?
References
- Maude, S. L., Teachey, D. T., Porter, D. L. & Grupp, S. A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 125, 4017–4024 (2015).
- Milone, M. C. et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Mol. Ther. 17, 1453–1464 (2009).
- Chang, Z. L. et al. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 14, 317–324 (2018).
- Roybal, K. T. et al. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 167, 1–14 (2016).
- Roybal, K. T. et al. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164, 770–779 (2016).
- Cho, J. H. et al. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses Article Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 1–13 (2018). doi:10.1016/j.cell.2018.03.038
- Roth, T. L. et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409 (2018).
doi: https://doi.org/10.1242/prelights.11394
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioengineering category:
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Detergent-Triggered Membrane Remodelling Monitored via Intramembrane Fluorescence De-Quenching
Cyntia Alves Conceição, Marcus Oliveira
A Novel Chimeric Antigen Receptor (CAR) - Strategy to Target EGFRVIII-Mutated Glioblastoma Cells via Macrophages
Dina Kabbara
Also in the cancer biology category:
Targeting CXADR-mediated AKT signaling suppresses tumorigenesis and enhances chemotherapy efficacy in Ewing sarcoma
TheLangeLab et al.
Dual inhibition of GTP-bound (ON) and GDP-bound (OFF) KRASG12C suppresses PI3Kα and leads to potent tumor inhibition
Luis Luna Ramírez
Taxane-Induced Conformational Changes in the Microtubule Lattice Activate GEF-H1-Dependent RhoA Signaling
Vibha SINGH
preLists in the bioengineering category:
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
3D Gastruloids
A curated list of preprints related to Gastruloids (in vitro models of early development obtained by 3D aggregation of embryonic cells). Updated until July 2021.
| List by | Paul Gerald L. Sanchez and Stefano Vianello |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Advances in microscopy
This preList highlights exciting unpublished preprint articles describing advances in microscopy with a focus on light-sheet microscopy.
| List by | Stephan Daetwyler |
Also in the cancer biology category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Anticancer agents: Discovery and clinical use
Preprints that describe the discovery of anticancer agents and their clinical use. Includes both small molecules and macromolecules like biologics.
| List by | Zhang-He Goh |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |