








A Phosphoproteomics Data Resource for Systems-level Modeling of Kinase Signaling Networks
Posted on: 30 August 2023 , updated on: 31 August 2023
Preprint posted on 3 August 2023
Discovering the dynamic world of the EGFR-MAPK phosphoproteome: Feng, Sanford and colleagues developed a multiplexed deep phosphoproteome profiling workflow unveiling 4500 protein sites exhibiting increased phosphorylation upon EGF stimulation.
Selected by Benjamin Dominik MaierCategories: bioinformatics, molecular biology, systems biology
Background
EGFR-MAPK signalling pathway
EGFR/MAPK signalling is one of the most studied signalling pathways and regulates various cellular processes such as cell growth, proliferation, and differentiation (Oda et al., 2005; Wee & Wang, 2017). Upon activation of the signalling pathway by epidermal growth factors (EGF) binding to epidermal growth factor receptors (EGFR) on the cell surface, the extracellular signal is transmitted and amplified within the cell through second messengers and cascades of phosphorylation events. Governed by kinases (adding phosphate groups to specific amino acids residues) and phosphatases (removing them), protein phosphorylation serves as a molecular on/off-switch regulating the activity, localization, and interaction of proteins through conformational changes (Ardito et al., 2017). Ultimately, this activates transcription factors and effector proteins triggering alterations in gene expression, enzyme activity, or induction of cell death.
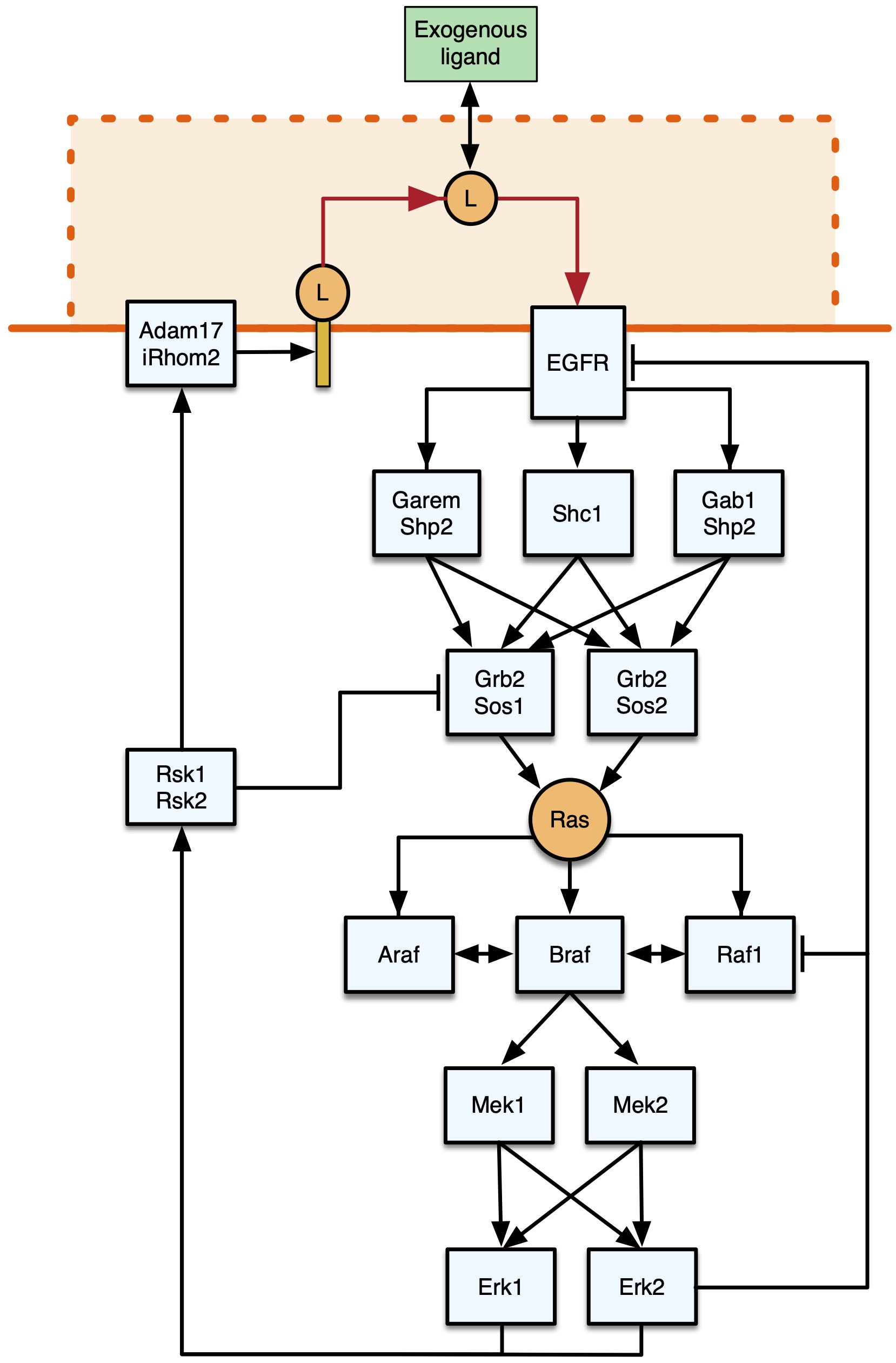
Fig. 1 Representation of the core EGFR-MAPK signalling pathway. Figure taken from Feng, Sangford et al. (2023), BioRxiv published under the CC-BY-NC-ND 4.0 International licence.
In healthy tissue, feedback mechanisms tightly regulate signalling to prevent excessive reactions and maintain cellular balance (Lemmon et al., 2016). If dysregulated, EGFR signalling contributes to various diseases, including cancer, inflammation, vascular diseases, and Alzheimer’s (Wieduwilt and Moasser, 2008). Consequently, gaining a comprehensive understanding of how genetic alterations contribute to dysregulation and discovering means to restore normal function becomes paramount for developing targeted therapies.
Phosphoproteome Profiling and High-throughput perturbational datasets
Protein phosphorylations are usually quantified through mass spectrometry-based methods (Yu & Veenstra, 2021). Common workflows involve ionising phosphopeptides, separating them based on their mass-to-charge ratio, and measuring their abundance in a sample. Recently, multiplexing methods using isobaric tags have been developed (e.g. Mertins et al., 2018), which enable simultaneous analysis of multiple samples over time and various doses in a single assay minimising variability and noise between samples.
The effects of chemical or genetic perturbations on cellular signalling can be studied using automated cost-effective transcriptomics and image-based profiling technologies which resulted in the extensive public JUMP Cell Painting (Chandrasekaran et al., 2023) and L1000 Connectivity Map (Subramanian et al., 2017) data sets. For more details, please check out my recent preLight posts on “Phospho-seq” and “Similarity metric learning on perturbational datasets”.
Mathematical Models
As it is impossible to experimentally study all possible biological contexts, researchers employ mechanistic mathematical models to investigate cell signalling. By combining literature with time and dosage-resolved experimental data, computational representations of molecular interactions and regulatory mechanisms can be created. Through simulations of a very large number of conditions and mathematical analysis, these models can help uncover the underlying principles governing cellular responses, prioritise which conditions are worth following up experimentally and aid in the design of targeted therapies. Yet, models inherently simplify reality and thus are not entirely accurate, often overlooking complex properties, lacking rigorous experimental validation, and/or being uninterpretable black-box models. Or in George Box’s words: “All models are wrong – but some are useful”.
Key Findings
Overview
Feng, Sanford and colleagues introduced a multiplexed phosphoproteome profiling technique to create an extensive dataset focused on the EGFR-MAPK pathway in non-transformed cells under physiological conditions. By integrating their data with various protein databases, the authors validated and expanded our understanding of EGFR-ERK pathway activation and feedback regulation. Their findings highlight biphasic signalling behaviours and unveil key regulatory components. In future, their data might be used to improve existing and construct novel mathematical models of EGFR-MAPK signalling and their downstream effects.
Comprehensive Phosphoproteomics Dataset
First, the authors determined optimal treatment conditions across time, EGF dosage and inhibitor dosage using enzyme-linked immunoassays (ELISA) to measure RAS and MAPK activity. Unlike earlier research, they used a non-cancerous cell line and EGF dosages that mimic human physiology. Their study focused solely on the EGFR-MAPK signalling cascade, ignoring delayed cellular responses like EGF-induced gene expression or protein turnover. Their initial results were found to be in line with previous studies and could be reproduced in computational simulations.
Following up on this, the authors used tandem mass spectroscopy to create three detailed datasets on phosphoproteomics, covering time-series, dose-series, and inhibitor effects in response to EGF. The goal was to understand how specific sites on proteins undergo phosphorylation changes in response to EGF. However, tandem mass spectroscopy sometimes provided uncertain results about the exact phosphorylation site. Hence, they developed a computational method that uses confidence values from a phosphosite predictor and the PhosphositePlus database information to accurately identify the correct phosphorylation sites. This method improved the accuracy of mapping phosphosites and ensured better alignment of their data with existing scientific literature. Next, they recalculated the intensity values of multi-phosphorylated phosphopeptides to obtain individual values for each phosphosite simplifying comparisons with prior literature and reducing complexity.
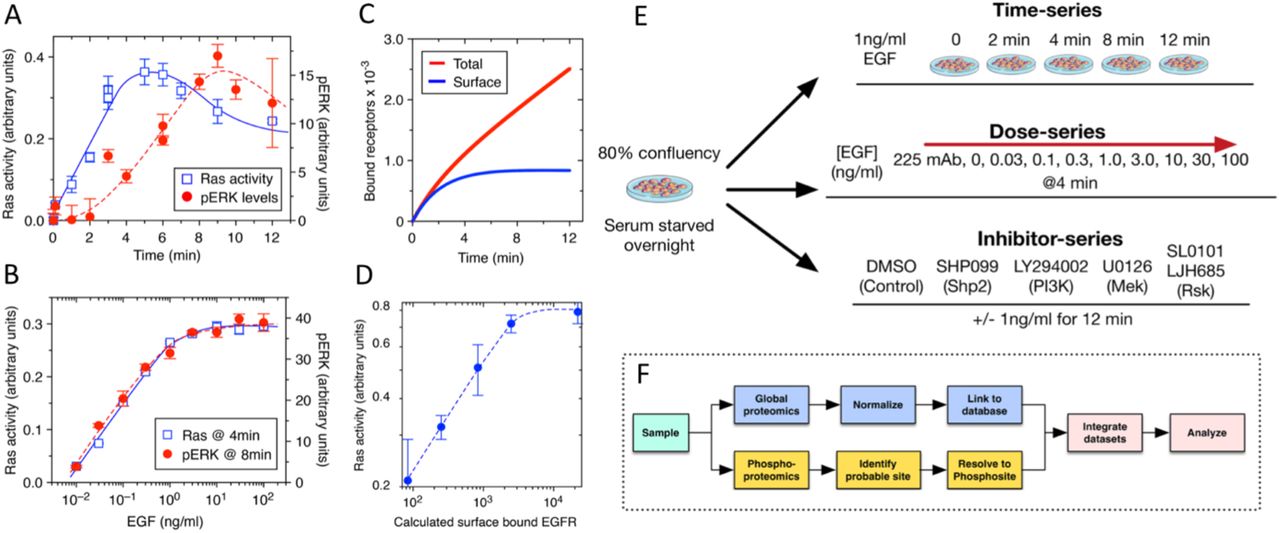
Fig. 2 Experimental Setup. Figure taken from Feng, Sangford et al. (2023), BioRxiv published under the CC-BY-NC-ND 4.0 International licence.
Simultaneously, the global protein abundance was measured and normalised across samples to rule out that observed changes in enzyme activity were caused by changes in protein expression instead of changes in phosphorylation.
To enhance the usability of the phosphoproteomics data, the measured data was combined with publicly available protein-specific information known to be relevant for modelling signalling pathways. This included abundance, interactions, localization, and functional roles. This resulted in an open-access resource called Phosphoprotein Explorer, containing 46,000+ phosphorylation sites on 6,600 proteins. Notably, approximately 4,500 sites from 2,110 proteins were found to be significantly enriched in response to EGF stimulation.
EGFR-MAPK Pathway Complexity
To assess the quality and resolution of the phosphoproteomics data, the authors constructed a literature-derived model of the EGF-induced MAPK pathway with known phosphorylation effects. They found that their phosphoproteomics data aligns well with known phosphorylation dynamics both for positive and negative phosphorylation events at relevant timescales and even for low abundance species.
In a more detailed analysis, they constructed systems-level maps for the EGFR-activated phosphorylation network using their extensive experimental dataset along with external protein data. Incorporating inhibitor data and PhosphoSitePlus references, they unravelled the network’s topology, including positive and negative feedback phosphorylation among crucial EGFR-MAPK pathway proteins. 18 proteins with 41 notable phosphorylation changes were identified in response to EGF, with 29 linked to activation and 12 to inhibition.
Next, the analysis was extended to include downstream proteins of RAS and MAPK, focusing on those with a minimum 2-fold increase in phosphorylation across experiments. As earlier studies concluded that key regulatory proteins are usually of low abundance and display a high number of phosphorylation sites, the list was further filtered accordingly yielding 29 proteins. While half of these proteins were already recognized as significant, the previously unidentified ones exhibited high functional scores, implying their newfound importance.
Conclusion and Perspective
While it feels challenging to keep track of relevant literature in the phosphoproteomics world, it is exciting to do research with all these new experimental and computational methods as well as access to new extensive datasets. For instance, in the same week as this article, a new article on the detection of post-translational modifications within long polypeptides by nanopore technology got published as well as a new preprint on a machine learning method to build time-resolved, functional phosphosignaling networks.
What I really like about the manuscript I feature in this preLights post is that the work is both experimental and computational and that it is only possible due to recent advances in both domains. Moreover, I believe that the created resource will be of great use for the community as it is a comprehensive dataset at relevant time scales and under physiological conditions. While writing the preLights post, Steven Wiley (senior author) guided me through the new database. The database is divided into gene/protein information and specific phosphorylation sites, with external proteomics database links for comprehensive protein details. Steve demonstrated the remarkable exploration potential of this tool by utilizing its highly adaptable search function, allowing the construction of intricate queries across all data fields. Currently, the tool contains the response data of MCF10A cells to EGF (this study), but the authors plan to consistently integrate new information and links as they emerge. Personally, I really look forward to potentially using the dataset to either validate or refine my current mechanistic EGFR model.
References
Ardito, F., Giuliani, M., Perrone, D., Troiano, G., & Lo Muzio, L. (2017). The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). International journal of molecular medicine, 40(2), 271–280. https://doi.org/10.3892/ijmm.2017.3036
Chandrasekaran, S. N., Ackerman, J., Alix, E., Ando, D. M., Arevalo, J., Bennion, M., Boisseau, N., Borowa, A., Boyd, J. D., Brino, L., Byrne, P. J., Ceulemans, H., Ch’ng, C., Cimini, B. A., Clevert, D.-A., Deflaux, N., Doench, J. G., Dorval, T., Doyonnas, R., … Carpenter, A. E. (2023). JUMP Cell Painting dataset: morphological impact of 136,000 chemical and genetic perturbations. Cold Spring Harbor Laboratory. https://doi.org/10.1101/2023.03.23.534023
Kalyuzhnyy, A., Eyers, P. A., Eyers, C. E., Bowler-Barnett, E., Martin, M. J., Sun, Z., Deutsch, E. W., & Jones, A. R. (2022). Profiling the Human Phosphoproteome to Estimate the True Extent of Protein Phosphorylation. Journal of proteome research, 21(6), 1510–1524. https://doi.org/10.1021/acs.jproteome.2c00131
Lemmon, M. A., Freed, D. M., Schlessinger, J., & Kiyatkin, A. (2016). The Dark Side of Cell Signaling: Positive Roles for Negative Regulators. Cell, 164(6), 1172–1184. https://doi.org/10.1016/j.cell.2016.02.047
Mertins, P., Tang, L.C., Krug, K. et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography–mass spectrometry. Nat Protoc 13, 1632–1661 (2018). https://doi.org/10.1038/s41596-018-0006-9
Ochoa, D., Jarnuczak, A. F., Viéitez, C., Gehre, M., Soucheray, M., Mateus, A., Kleefeldt, A. A., Hill, A., Garcia-Alonso, L., Stein, F., Krogan, N. J., Savitski, M. M., Swaney, D. L., Vizcaíno, J. A., Noh, K. M., & Beltrao, P. (2020). The functional landscape of the human phosphoproteome. Nature biotechnology, 38(3), 365–373. https://doi.org/10.1038/s41587-019-0344-3
Oda, K., Matsuoka, Y., Funahashi, A., & Kitano, H. (2005). A comprehensive pathway map of epidermal growth factor receptor signaling. Molecular systems biology, 1, 2005.0010. https://doi.org/10.1038/msb4100014
Subramanian, A., Narayan, R., Corsello, S. M., Peck, D. D., Natoli, T. E., Lu, X., Gould, J., Davis, J. F., Tubelli, A. A., Asiedu, J. K., Lahr, D. L., Hirschman, J. E., Liu, Z., Donahue, M., Julian, B., Khan, M., Wadden, D., Smith, I. C., Lam, D., Liberzon, A., … Golub, T. R. (2017). A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell, 171(6), 1437–1452.e17. https://doi.org/10.1016/j.cell.2017.10.049
Wee, P., & Wang, Z. (2017). Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers, 9(5), 52. https://doi.org/10.3390/cancers9050052
Wieduwilt, M. J., & Moasser, M. M. (2008). The epidermal growth factor receptor family: biology driving targeted therapeutics. Cellular and molecular life sciences : CMLS, 65(10), 1566–1584. https://doi.org/10.1007/s00018-008-7440-8
Yu, L. R., & Veenstra, T. D. (2021). Characterization of Phosphorylated Proteins Using Mass Spectrometry. Current protein & peptide science, 22(2), 148–157. https://doi.org/10.2174/1389203721999201123200439
doi: https://doi.org/10.1242/prelights.35317
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the bioinformatics category:
A potential anti-amyloidogenic therapy for type 2 diabetes based on the QBP1 peptide
Joao Gabriel, Marcus Oliveira
The lipidomic architecture of the mouse brain
CRM UoE Journal Club et al.
Kosmos: An AI Scientist for Autonomous Discovery
Roberto Amadio et al.
Also in the molecular biology category:
Detergent-Triggered Membrane Remodelling Monitored via Intramembrane Fluorescence De-Quenching
Cyntia Alves Conceição, Marcus Oliveira
Classical enhancers couple cis-regulatory logic with transcriptional condensates and 3D genome architecture
Siddharth Singh
Small Molecule Agonists of TREM2 Reprogram Microglia and Protect Synapses in Human Alzheimer’s Models
Dina Kabbara
Also in the systems biology category:
Human single-cell atlas analysis reveals heterogeneous endothelial signaling
Charis Qi
Longitudinal single cell RNA-sequencing reveals evolution of micro- and macro-states in chronic myeloid leukemia
Charis Qi
Environmental and Maternal Imprints on Infant Gut Metabolic Programming
Siddharth Singh
preLists in the bioinformatics category:
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the molecular biology category:
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |
Also in the systems biology category:
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Pattern formation during development
The aim of this preList is to integrate results about the mechanisms that govern patterning during development, from genes implicated in the processes to theoritical models of pattern formation in nature.
| List by | Alexa Sadier |