








Disrupting Transcriptional Feedback Yields an Escape-Resistant Antiviral
Posted on: 3 January 2019
Preprint posted on 26 November 2018
Article now published in Cell at http://dx.doi.org/10.1016/j.cell.2022.04.022
Dr. Weinberger and colleagues create a new anti-viral drug modality that uses a 28 base pair piece of DNA to have the virus reduce its own progression without any sign of resistance.
Selected by Pavithran RavindranCategories: biochemistry, microbiology, molecular biology
Background
From cancers to viruses, treatment of diseases is hindered by the emergence of resistance. Resistance is typically thought to arise by mutations in the protein that a drug is targeting. Diseases such as cancer and viruses have very high mutation rates and thus resistance tends to crop up quite quickly. Therefore, scientists have tried to utilize combination drug protocols that target multiple nodes in a particular disease development (e.g. the reverse transcriptase and integrase of a virus). There are two problems with such a method: (a) there must be two distinct drug targets available and (b) the drugs for both targets need to have compatible bioavailability, kinetics, efficacy, toxicity, etc. This scheme has been proving effective, but still does not seem to work, for resistance still occurs. The authors of this work wanted to find a new modality by which viral infections could be targeted and, ideally, cured without resistance.
Key Findings
One target that has not been exploited as of yet is auto-regulatory circuits that are present in viruses, specifically in herpes simplex virus type one (HSV-1) and cytomegalovirus (CMV). In normal viral progression, proteins such as IE86 in CMV and IE175 in HSV-1 are very closely regulated by negatively feeding back on their own promoters. However, if this circuit were to be broken, these proteins would build up in large amounts, kill the host cell, and ultimately decrease viral progression. The authors of this study chose to make a new type of oligonucleotide “drug” that would mimic the domain of the promoter that these IE proteins bind to, titrate away the IE proteins from the promoter they should be acting on and thus deregulate the amount of this cytotoxic protein that is made. They called this new drug modality circuit disruptor oligonucleotide therapy (C-DOTs). With this, infected cells would die and reduce the viral progression (Figure 1).
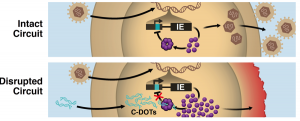
The authors first had to find an oligonucleotide sequence that could bind the IE proteins and titrate them away from their natural binding partner. For this, they developed a liquid chromatography based assay that could detect IE protein, DNA and the complex of the two together. With this assay they determined that the optimal oligonucleotide size is 28 base pairs. They further validated some of their hits using a GFP attached to the IE86 protein that was driven by the promoter for IE86 that should be shut down once some IE86 is made. When these cells were treated with the 28 base pair oligonucleotide matching the cis-regulatory sequence in the promoter, there was a significant increase in GFP positive cells.
To evaluate C-DOTS for anti-viral activity, the authors took the C-DOT against CMV (C-DOTC) and against HSV-1 (C-DOTH) and treated cells that were infected with the corresponding virus. The authors were able to find something especially interesting – at low multiplicity of infection (MOI) the C-DOT treatment worked as well as the conventional therapy but at higher MOI the C-DOTs worked better. This is not surprising when one considers the fact that the C-DOT therapy takes advantage of increased amounts of the IE proteins that should be regulated closely; in the case of the higher MOI, there are more viral genomes per cell and thus, more opportunities to have increased amount of IE protein to cause cell death and decreased viral load. Amazingly, the authors found that this treatment could be used for a wide range of viral strains, not just the one that they were using to characterize the treatment in the first place.
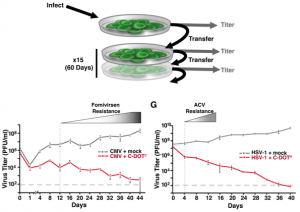
The biggest concern that the authors were trying to combat was resistance. To test if the C-DOT therapy could do better than conventional therapies (Fomivirsen for CMV and ACV for HSV-1) in terms of resistance strains forming, the authors performed an assay in which the virus was continuously passaged from infected cells to non-infected cells every four days and the virus was tittered during each passaging. They found that the conventional therapies produced resistance within 12 days while the C-DOT therapy amazingly had essentially zero resistance form for up to 40 days in culturing cells, with CMV titers staying below the limit of detection at day 52 (Figure 2). The authors were even able to take this to the next level by implementing their treatment in mice. When C-DOTs were given to mice challenged with the corresponding virus, the authors saw amazing reduction in viral loads (150 fold decrease in the case of HSV-1).
Why I chose this preprint
The simplicity yet brilliance of the work described here is the major reason why I chose this particular preprint- the idea that one can use the viruses’ own regulatory networks to get it to kill itself. This was simply done by titrating away the DNA binding protein using small oligonucleotides. Not only does this work provide a very promising therapy for CMV and HSV-1 described here, but it also provides a new therapeutic modality to test for other diseases such as cancer. A lot of work has been done showing how cancers rewire regulatory networks and so being able to take advantage of that would be an amazing achievement [1-2]. Furthermore, the readability of this particular work made it an especially fun reading experience – the language was concise and the figures were very clear with panels explaining hypothesized results.
Outstanding Questions
- What is the difference between Fomivirsen and the newer C-DOTC? How does the resistance arise for Fomivirsen and does this mechanism apply to C-DOTC?
- Is there a way that the binding of the C-DOT and IE protein can be compared to the endogenous promoter? If so, is there a way to increase the C-DOT binding to IE protein such that it is more preferential compared to the endogenous location?
- The idea that such feedback loops can be targeted for not just viruses but also other diseases such as cancers is very attractive. However, one of my questions is how one can identify targetable feedback loops?
References
- Fish L, Zhang S, Yu JX, et al. Cancer cells exploit an orphan RNA to drive metastatic progression. Nat Med. 2018;24(11):1743-1751.
- Bugaj LJ, Sabnis AJ, Mitchell A, et al. Cancer mutations and targeted drugs can disrupt dynamic signal encoding by the Ras-Erk pathway. Science. 2018;361(6405).
doi: https://doi.org/10.1242/prelights.6718
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the biochemistry category:
Site-Specific Inhibition of Translation Initiation via 2’-O-methylation
Leonie Brüne
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
Also in the microbiology category:
Circadian Clock Programming of Anticipatory Antiviral Immunity Gates Enteric Virus Infection Susceptibility
Owen Ang
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
Gut microbiome changes over the course of multiple sclerosis differentially influence autoimmune neuroinflammation
Carole Djagang et al.
Also in the molecular biology category:
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Defective BRCA1-mediated DNA end resection drives tandem duplication formation and FANCM synthetic lethality
Marta San Martin
Thousandfold Expansion Microscopy
Felipe Del Valle Batalla
preLists in the biochemistry category:
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
Peer Review in Biomedical Sciences
Communication of scientific knowledge has changed dramatically in recent decades and the public perception of scientific discoveries depends on the peer review process of articles published in scientific journals. Preprints are key vehicles for the dissemination of scientific discoveries, but they are still not properly recognized by the scientific community since peer review is very limited. On the other hand, peer review is very heterogeneous and a fundamental aspect to improve it is to train young scientists on how to think critically and how to evaluate scientific knowledge in a professional way. Thus, this course aims to: i) train students on how to perform peer review of scientific manuscripts in a professional manner; ii) develop students' critical thinking; iii) contribute to the appreciation of preprints as important vehicles for the dissemination of scientific knowledge without restrictions; iv) contribute to the development of students' curricula, as their opinions will be published and indexed on the preLights platform. The evaluations will be based on qualitative analyses of the oral presentations of preprints in the field of biomedical sciences deposited in the bioRxiv server, of the critical reports written by the students, as well as of the participation of the students during the preprints discussions.
| List by | Marcus Oliveira et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Cellular metabolism
A curated list of preprints related to cellular metabolism at Biorxiv by Pablo Ranea Robles from the Prelights community. Special interest on lipid metabolism, peroxisomes and mitochondria.
| List by | Pablo Ranea Robles |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |
Also in the microbiology category:
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
BioMalPar XVI: Biology and Pathology of the Malaria Parasite
[under construction] Preprints presented at the (fully virtual) EMBL BioMalPar XVI, 17-18 May 2020 #emblmalaria
| List by | Dey Lab, Samantha Seah |
1
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Antimicrobials: Discovery, clinical use, and development of resistance
Preprints that describe the discovery of new antimicrobials and any improvements made regarding their clinical use. Includes preprints that detail the factors affecting antimicrobial selection and the development of antimicrobial resistance.
| List by | Zhang-He Goh |
Also in the molecular biology category:
Developmental regulation: molecular and ecological niches
This conference was held at the Station Biologique de Roscoff (France) and brought together researchers exploring how diverse niche environments shape developmental processes across scales. Spanning topics from ecological and metabolic influences to signalling networks, mechanics and gene regulation, the meeting highlighted the interplay between intrinsic and extrinsic factors in controlling cell fate and tissue organisation. This preList gathers preprints discussed by speakers and poster presenters during the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |