








Endogenous retroviruses are a source of enhancers with oncogenic potential in acute myeloid leukaemia
Posted on: 21 April 2020
Preprint posted on 4 April 2020
Article now published in Nature Communications at http://dx.doi.org/10.1038/s41467-020-17206-4
Tumours take advantage of transposons: endogenous retroviruses can act as enhancers in acute myeloid leukemia.
Selected by Jesus VictorinoCategories: cancer biology, genomics
Background & Summary
Between one half to two thirds of the human genome consists of repetitive sequences, most of which are inactivated transposable elements (TEs). These sequences of viral origin integrated into our genomes along evolution. The enormous proportion of TEs suggests that they have played an important role in shaping our genome [1]. Given that the ENCODE Project estimates that at least 80% of the entire genome has functional annotations –such as active transcription, histone marks or transcription factor (TF) binding [2]– just by doing the math one could think that such DNA ‘fossils’ likely contain some functionality.
TEs can act as cis-regulatory modules controlling gene expression although they are epigenetically silenced during early development [3]. Several studies have shown deregulation of TEs, including endogenous retroviruses (ERVs), in different types of cancer. In this preprint, Deniz and collaborators explore the pathogenic role of TEs in acute myeloid leukemia (AML).
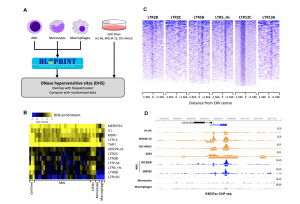
Figure 1. Activation of ERVs with regulatory potential in AML. A) Obtention of DNase-seq data for differentiated cells (monocytes and macrophages), hematopoietic stem cells and AML (both from primary samples and cell lines). B) Heatmap showing that chromatin is specifically open at several families of ERV. C) DNase-seq profiles in AML per family of ERV. D) Example of an ERV element behaving as an AML-specific enhancer. This figure has been modified from Figures 1 and 2 from the original preprint.
Key findings
– AML cells activate several families of ERVs. AML cells coming from both primary samples and established cell lines shared enrichment of open chromatin and enhancer signatures as well as binding of leukemia-related TFs in six families of ERVs (Fig. 1). This is in stark contrast to what happens in differentiated blood cells, such as macrophages and monocytes which do not activate these families (Fig. 1B).
– Individual ERV enhancers regulate gene expression. ERVs containing signatures of open chromatin correlated with higher expression of close genes. Deletion of individual ERV candidates proved their enhancer activity by downregulation of specific genes (Fig. 2A and B).
– Broad epigenomic silencing of ERVs reduced cell proliferation. The authors leveraged the repetitive nature of these TEs to target more than 200 of these sites by CRISPR interference (CRISPRi), a sequence-specific tool for gene silencing. Epigenomic editing altered the epigenomic landscape of targets and reduced proliferation in leukemia cells. The downregulated genes APOC1 or APOE seemed to impair cell growth thus contributing to ameliorate the malignant phenotype (Fig. 2C, D and E).
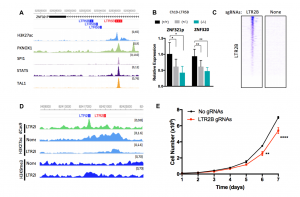
Figure 2. Perturbation of enhancers at ERVs regulates gene expression and affects proliferation. A) LTR5B element at an intron of ZNF321P shows open chromatin and TF binding. B) Deletion of the ERV in A downregulate target genes ZNF321P and ZNF320. C) dCas9-KRAB binds to hundreds of LTR2B elements targeting epigenomic silencing (CRISPRi). D) CRISPRi effectively edit the epigenomic landscape at target sites. E) Downregulation of target genes mediated by CRISPRi leads to impaired proliferation. This figure has been modified from Figures 4 and 5 from the original preprint.
Why I like this preprint
Considering the epigenomic status of cells and tissues as an accurate footprint of their identity and behaviour could be “too simple, sometimes naïve”. However, there is increasing evidence of diseases and disorders with common chromatin-related features such as enhancer rewiring or changes in global DNA methylation or post-translational modifications of histones. Since we are still not able to understand most of the information that global genetic and epigenetic analysis from patients provide it is very important to find disease hallmarks in order to benefit diagnosis and treatment strategies.
This work links two very interesting questions: ‘what is the epigenetic footprint of leukemia cells?’ and ‘what does a huge and repetitive part of the genome have to do with that?’. I like this preprint because the authors provide a combined answer to these questions and turn a potential genetic limitation such as working with very similar sequences into an interesting approach to study global effects of epigenetic silencing.
Question to the authors
– The broad CRISPRi-mediated repression performed in LTR2B elements led to epigenetic changes at target loci and downregulation of several genes. However, the ERV element located between the genes APOC1 and APOE seems to be the responsible for the proliferative phenotype. Do the authors think that the rest of the elements targeted through this approach (especially those downregulating their target genes) also contribute to clonal advantages in AML? And, if so, how?
– How do the authors show that downregulation of APOC1 gene and not that of APOE is responsible for the proliferative phenotype? Is any of those gene consistently upregulated in most AML samples?
– The authors showed that the intronic LTR5b element located at ZNF321P gene regulate its expression as well as that of ZNF320 and ZNF888 genes. How do the authors explain downregulation of ZNF320 (Figure 5F) after the CRISPRi-mediated repression targeting LTR2B elements? Do the authors also see downregulation of ZNF321P and ZNF888?
– Have the authors assess the effect in proliferation of other genes downregulated, such as ZNF320, in the CRISPRi experiment?
References
- de Koning AJ et al. 2011. Repetitive Elements May Comprise Over Two-Thirds of the Human Genome. PLoS Genet. 7(12):e1002384.
- ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74.
- Chuong EB et al. 2017. Regulatory activities of transposable elements: from conflicts to benefits. Nature Rev Genet. 18, 71-86.
doi: https://doi.org/10.1242/prelights.19000
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the cancer biology category:
Targeting CXADR-mediated AKT signaling suppresses tumorigenesis and enhances chemotherapy efficacy in Ewing sarcoma
TheLangeLab et al.
Dual inhibition of GTP-bound (ON) and GDP-bound (OFF) KRASG12C suppresses PI3Kα and leads to potent tumor inhibition
Luis Luna Ramírez
A Novel Chimeric Antigen Receptor (CAR) - Strategy to Target EGFRVIII-Mutated Glioblastoma Cells via Macrophages
Dina Kabbara
Also in the genomics category:
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
preLists in the cancer biology category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Anticancer agents: Discovery and clinical use
Preprints that describe the discovery of anticancer agents and their clinical use. Includes both small molecules and macromolecules like biologics.
| List by | Zhang-He Goh |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |
Also in the genomics category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |