








Reconstruction of the global neural crest gene regulatory network in vivo
Posted on: 28 January 2019
Preprint posted on 30 December 2018
Article now published in Developmental Cell at http://dx.doi.org/10.1016/j.devcel.2019.10.003
How to build a vertebrate neural crest: reconstructing neural crest gene regulatory networks in vivo
Selected by Hannah BrunsdonCategories: developmental biology, genomics
Introduction
Gene regulatory networks (GRNs) are formed of molecular regulators encoded in DNA, which process information from transcription factors and signalling molecules to regulate many growth and developmental processes. Much of what we know about vertebrate GRNs is inferred from manipulation of candidate genes, and GRNs of simpler organisms – the sheer complexity of vertebrate gene regulation makes it extremely difficult to gain an unbiased view of GRN interactions in vertebrate development. This has not deterred the authors of a recent preprint from the Sauka-Spengler lab, however. Their work shows the output of an enormous effort in reconstructing the chick neural crest (NC) GRN using both transcriptomic and epigenomic profiling, thus creating the first high-resolution, genome-wide reconstruction of a vertebrate GRN in vivo.
Key findings
To gain an overview of the early cranial NC transcriptional landscape, the authors isolated NC cells from chick embryos at both pre-migratory stages (5-6ss) and early migratory stages (8-10ss) for RNA seq. The data clustered into 13 gene expression modules that are associated with the changing nature of the NC as cells undergo EMT and delaminate from the neural tube. To investigate the regulation of these processes at the epigenomic level, the authors performed ATAC-seq in conjunction with Next-Generation Capture-C on stage-matched samples. This revealed differences in chromatin accessibility upstream of core NC genes, revealing the presence of enhancer elements, as well as physical interactions between these and putative target genes.
Diving deeper into the ATAC-seq data, the authors performed k-means clustering analyses based on patterns of chromatin accessibility. Of particular interest was “k-cluster 3”, which contained elements only accessible in NC cells, and “k-cluster 9”-containing elements, enriched in both the NC and neighbouring neuroepithelial ectoderm. Cis-regulatory elements from both these clusters were then assigned to their nearest-neighbour genes, in order to build predictive networks of how different combinations of active enhancers cooperate in NC gene activation.
What I think makes this preprint so special, is that predictions made from their different datasets are tested and visualised in vivo. For example, the locus of Sox10 – a known master regulator of NC cell fate – was investigated by ChIP-seq to find novel putative enhancers. The activity of these enhancers was visualised in vivo by electroporating chick embryos with fluorescently-tagged enhancer reporter constructs. Individual Sox10 enhancer activity was spatially and temporally diverse, showing complex cis-regulation of its expression, as seen below:
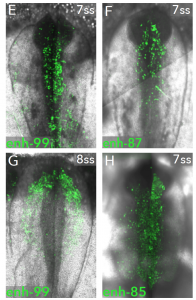
From preprint Figure 4. In vivo activity of novel Sox10 enhancers at the 7 and 8 somite stage.
Taking this a step further, CRISPR-mediated genome editing was employed to test the functionality of these Sox10 enhancers. Guide RNAs targeting each enhancer were electroporated into chick embryos alongside the transcriptional repressor dCas9-Krab, thus selectively disrupting their activity. Subsequent RT-qPCR analysis on isolated neural tubes showed that disruption of single enhancers did not affect Sox10 expression for long, but simultaneous repression of certain enhancer combinations caused long-term repression.
So, does this heterogeneity of cis-regulatory elements influence NC lineage decisions? GO term enrichment analysis was performed on genes associated to k-cluster 3 and 9 elements. This confirmed that open k-cluster 3 elements drive NC development and differentiation programmes, whereas open k-cluster 9 elements prime cells for more neural fates. To investigate if these differences produce meaningful transcriptomic changes within individual NC cells, single cell RNA-seq was performed at the point of NC delamination. PCA analysis of the data revealed three gene clusters. Sc-cluster 1 and 3 strongly overlapped with k-cluster 3 genes and were characteristic of the canonical NC, whereas sc-cluster 2 was more characteristic of NC-derived neural progenitors, and strongly correlated with k-cluster 9 genes. Therefore, well before commitment of cells to distinct NC-derived lineages, there is already considerable heterogeneity in regulatory elements, which must be important for establishing diverse NC progenitor lineages.
In the final part of their paper, the authors identified transcription factor co-binding sites within their integrated transcriptomic and epigenomic data, thus building a comprehensive, genome-wide reconstruction of k-cluster 3 and k-cluster 9-dependent GRNs, and identifying the hierarchies of transcription factors that comprised each GRN’s minimal regulatory cores. Through CRISPR-Cas9-mediated disruption of these minimal cores and subsequent RNA-seq, the authors confirmed that the balance of k-cluster 3 and 9 GRNs, with likely cross-repressive interactions, regulates the balance between NC-derived neural and mesenchymal lineages.
If it wasn’t already clear, this research has generated a huge amount of data, tying together transcriptional, epigenetic, single-cell and bulk RNA-seq experiments, as well as in vivo validation. Thankfully for neural crest enthusiasts unfamiliar with manipulating large datasets, the authors have assembled their data into a user-friendly Shiny App (R-based, available from GitHub). Below are screenshots of the App interface visualising the upstream regulatory elements and k-cluster-dependent transcriptional networks of TFAP2B:
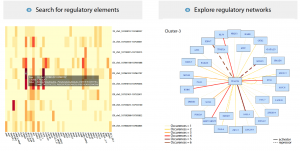
From Figure 7 in the preprint. Images from the Shiny App showing the regulatory elements, and transcription factor networks for TFAP2B
Why I chose this preprint
This preprint represents an incredible amount of work and team effort, and I was amazed by the number of Next-Generation seq analyses that were performed throughout the study, but nevertheless effectively pulled together to build a cohesive story about the early NC GRN. Secondly, whereas other large scale transcriptomics/epigenomics studies sometimes conclude with their computational predictive models, this preprint matches their intricate dataset analyses with ambitious in vivo work too. For people like myself, who are more comfortable with wet lab experiments than manipulating huge datasets, the provision of a friendly app interface to enable the research community to play with the data themselves is a very valuable resource.
Questions for the authors
- Given the complexity of the paper, this question will probably now sound really simplistic, but what is it about the chick as a model organism that made it ideal for building NC GRNs?
- Do you have any plans to investigate other k-clusters and developmental stages to compare their GRNs to k-cluster 6 and 9? It would be interesting to know how k-cluster 6, which is involved in early establishment of the NC could differ to, and interact with, k-cluster 3 and 9 for example. Following on from that, by sampling at other stages would it be possible to follow NC GRNs from NC establishment to final lineage differentiation in this way?
doi: https://doi.org/10.1242/prelights.7943
Read preprint (No Ratings Yet)
(No Ratings Yet)Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the developmental biology category:
Disordered protein COSA-2 maintains crossover-specific repair compartments to ensure meiotic crossover maturation
Chee Kiang Ewe
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Developmental conversion of the nucleolus into an RNA Polymerase II transcriptional platform in Drosophila spermatocytes
Panagiotis Giannios
Also in the genomics category:
Comprehensive Lineage Tracing Maps the Landscape of Cell Fate Decisions in Mouse Embryogenesis
Béryl Laplace-Builhé, Lucie Hermet
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
preLists in the developmental biology category:
Developmental regulation: molecular and ecological niches
This conference was held at the Station Biologique de Roscoff (France) and brought together researchers exploring how diverse niche environments shape developmental processes across scales. Spanning topics from ecological and metabolic influences to signalling networks, mechanics and gene regulation, the meeting highlighted the interplay between intrinsic and extrinsic factors in controlling cell fate and tissue organisation. This preList gathers preprints discussed by speakers and poster presenters during the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
BSDB/GenSoc Spring Meeting 2024
A list of preprints highlighted at the British Society for Developmental Biology and Genetics Society joint Spring meeting 2024 at Warwick, UK.
| List by | Joyce Yu, Katherine Brown |
GfE/ DSDB meeting 2024
This preList highlights the preprints discussed at the 2024 joint German and Dutch developmental biology societies meeting that took place in March 2024 in Osnabrück, Germany.
| List by | Joyce Yu |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
preLights peer support – preprints of interest
This is a preprint repository to organise the preprints and preLights covered through the 'preLights peer support' initiative.
| List by | preLights peer support |
The Society for Developmental Biology 82nd Annual Meeting
This preList is made up of the preprints discussed during the Society for Developmental Biology 82nd Annual Meeting that took place in Chicago in July 2023.
| List by | Joyce Yu, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
2nd Conference of the Visegrád Group Society for Developmental Biology
Preprints from the 2nd Conference of the Visegrád Group Society for Developmental Biology (2-5 September, 2021, Szeged, Hungary)
| List by | Nándor Lipták |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
Single Cell Biology 2020
A list of preprints mentioned at the Wellcome Genome Campus Single Cell Biology 2020 meeting.
| List by | Alex Eve |
Society for Developmental Biology 79th Annual Meeting
Preprints at SDB 2020
| List by | Irepan Salvador-Martinez, Martin Estermann |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
Planar Cell Polarity – PCP
This preList contains preprints about the latest findings on Planar Cell Polarity (PCP) in various model organisms at the molecular, cellular and tissue levels.
| List by | Ana Dorrego-Rivas |
Cell Polarity
Recent research from the field of cell polarity is summarized in this list of preprints. It comprises of studies focusing on various forms of cell polarity ranging from epithelial polarity, planar cell polarity to front-to-rear polarity.
| List by | Yamini Ravichandran |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
3D Gastruloids
A curated list of preprints related to Gastruloids (in vitro models of early development obtained by 3D aggregation of embryonic cells). Updated until July 2021.
| List by | Paul Gerald L. Sanchez and Stefano Vianello |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EDBC Alicante 2019
Preprints presented at the European Developmental Biology Congress (EDBC) in Alicante, October 23-26 2019.
| List by | Sergio Menchero et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
SDB 78th Annual Meeting 2019
A curation of the preprints presented at the SDB meeting in Boston, July 26-30 2019. The preList will be updated throughout the duration of the meeting.
| List by | Alex Eve |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Young Embryologist Network Conference 2019
Preprints presented at the Young Embryologist Network 2019 conference, 13 May, The Francis Crick Institute, London
| List by | Alex Eve |
Pattern formation during development
The aim of this preList is to integrate results about the mechanisms that govern patterning during development, from genes implicated in the processes to theoritical models of pattern formation in nature.
| List by | Alexa Sadier |
BSCB/BSDB Annual Meeting 2019
Preprints presented at the BSCB/BSDB Annual Meeting 2019
| List by | Dey Lab |
Zebrafish immunology
A compilation of cutting-edge research that uses the zebrafish as a model system to elucidate novel immunological mechanisms in health and disease.
| List by | Shikha Nayar |
Also in the genomics category:
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
Early 2025 preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) bioinformatics 2) epigenetics 3) gene regulation 4) genomics 5) transcriptomics
| List by | Chee Kiang Ewe et al. |
End-of-year preprints – the genetics & genomics edition
In this community-driven preList, a group of preLighters, with expertise in different areas of genetics and genomics have worked together to create this preprint reading list. Categories include: 1) genomics 2) bioinformatics 3) gene regulation 4) epigenetics
| List by | Chee Kiang Ewe et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Semmelweis Symposium 2022: 40th anniversary of international medical education at Semmelweis University
This preList contains preprints discussed during the 'Semmelweis Symposium 2022' (7-9 November), organised around the 40th anniversary of international medical education at Semmelweis University covering a wide range of topics.
| List by | Nándor Lipták |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
EMBL Conference: From functional genomics to systems biology
Preprints presented at the virtual EMBL conference "from functional genomics and systems biology", 16-19 November 2020
| List by | Jesus Victorino |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |