








Precisely control mitochondria with light to manipulate cell fate decision
Posted on: 14 June 2019
Preprint posted on 29 May 2019
Article now published in Biophysical Journal at http://dx.doi.org/10.1016/j.bpj.2019.06.038
Using optogenetics, the ‘method of the decade’, to depolarise mitochondria.
Selected by Amberley StephensCategories: biochemistry, cell biology, neuroscience
Background
The methods used to alter cellular pathways or investigate the role of a protein commonly include overexpression or knock-out of the gene of interest, chemical activation or blocking of a receptor/ion channel/protein-protein interaction. However, these methods can have unknown downstream effects. Optogenetics offers a potentially cleaner method to study certain mechanisms whereby a light-sensitive protein is utilised. These proteins consist of seven transmembrane domains that form ion channels which open and close upon light stimulation.
Results
The correct localisation signal is needed to target channelrhodopsin to mitochondria
Ernst et al., used optogenetics to study the role of depolarisation of mitochondria and its effect on mitophagy, (the ordered degradation of mitochondria). They first investigated different sequences for targeting the channelrhodopsin (Ch2R) to the mitochondria and found that only a mitochondrial leader sequence copied from a large ABCB10 ATPase binding cassette transporter was able to localise the Ch2R.
Channelrhodopsin can depolarise mitochondria
Depolarisation of distinct areas with a 475 nm LED laser lead to the ChR2 ion channels opening and 80% depolarisation of mitochondria in this region compared to unilluminated control areas (Figure 1). Depolarisation was measured using tetramethylrhodamine, methyl ester (TMRM) fluorescence which decreases as the inner mitochondrial membrane potential (ΔΨm) decreases and TMRM is released into the cell. Light levels had to be moderated as high stimulation (7 mW/mm2) lead to increased cell death of the control cells.
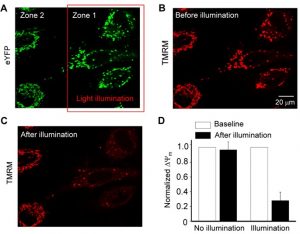
Figure 1, taken from preprint Figure 2. Mitochondrial-targeted optogenetics induces selective mitochondrial depolarization in H9C2 cells expressing ChR2-eYFP. A) Confocal image showing eYFP tagged ChR2 and cells in zone 1 which were illuminated by LED (5 mW/mm2 ) and cells in zone 2 were not exposed to illumination. B+C) Confocal images showing mitochondrial membrane potential (ΔΨm), measured by TMRM fluorescent dye (20 nM), of cells in zone 1 and zone 2 before (B) and after (C) light illumination. D) Normalized ΔΨm in cells in the illuminated (i.e. zone 1) and unilluminated (i.e. zone 2) zones before and after light illumination.
Sustained illumination leads to cell death via the apoptotic pathway
Moderate illumination (0.5 mW/mm2) for 24 hrs lead to cell death of the ChR2-eYFP cells, not the control cells. The authors subsequently investigated the mechanisms of this light-induced ChR2 mediated cell death. Cell death was found to be independent of mitochondrial permeability, transition pore opening and independent of reactive oxygen species production, as cell death was not prevented using inhibitors. An apoptosis and necrosis inhibitor were next tested and these experiments showed that the apoptosis inhibitor, which targets caspases, reduces cell death. This showed that light-induced ChR2 mediated cell death was via the apoptotic pathway.
Overexpression of Parkin induces mitophagy upon illumination
The authors then further investigated what occurs when mitochondria become damaged after sustained light activation by studying the PINK1/Parkin-mediated mitophagy pathway. When mitochondria become damaged the ΔΨm is disrupted and PINK1 can not be internalised for processing and is therefore left externally. Parkin, a ubiquitin ligase, recognises the externalised PINK1 and subsequently targets the mitochondria to the mitophagy pathway. Parkin was overexpressed in HeLa cells and over time (0-24 hrs) and under illumination (0.5 mW/mm2) there was an increase in Parkin and Ch2R-eYFP colocalization, an increase in mitochondrial fragmentation and a decrease in mitochondrial mass. This data, combined with the presence of the autophagosome marker, LC3, and lyososome marker, LysoTracker, indicated that mitophagy was occurring, which was not observed in control cells or unilluminated cells. Surprisingly, although pro-apoptotic effects of increased Parkin expression have been previously reported, the overexpression of Parkin did not lead to an increase in cell survival, which may have been expected due to damaged mitochondria being more efficiently removed.
Preconditioning of mitochondria can enhance cell viability
Corroborating previous studies which have shown that preconditioning of mitochondria can increase cell survival during sustained stress, the authors also observed that preconditioning with low-level illumination (2 hours for 0.2 mW/mm2) increased cell viability from ~40% without preconditioning to ~80% viability after illumination for 6 hours at 4 mW/mm2.
Conclusions
Optogenetics can be successfully used to depolarise mitochondria upon light-activation by directing channelrhodopsin to the mitochondrial inner membrane using a mitochondrial leader sequence from the ABCB10 transporter. This technique will allow targeted investigation into the role of the mitochondrial inner membrane potential (ΔΨm) under physiological conditions and under cell stress/death. Overexpression of Parkin leads to the induction of mitophagy with sustained illumination. Further investigation into the role of Parkin in mitophagy may yield potential ways to moderate it to reduce disease burden.
Why I chose this preprint
I am a great fan of techniques that circumvent the use of protein over-expression or chemical inhibition where we don’t really know what downstream effects may be perturbed in the cell. I also liked that the authors explained how they found a mitochondrial leader sequence that worked. There is often a lot of work that goes on behind the scenes which gets left out of papers that may be beneficial to know.
Questions for Authors
How do you know the ChR2 is definitely at the IMM?
Is it possible to block or mutate the ChR2 channel to determine whether it is definitely a transfer of protons through this channel causing depolarisation?
Can you speculate as to what mechanisms induce this protection of cell viability when mitochondria are preconditioned? Has anyone looked at this?
doi: https://doi.org/10.1242/prelights.11293
Read preprint (No Ratings Yet)
(No Ratings Yet)Have your say
Sign up to customise the site to your preferences and to receive alerts
Register hereAlso in the biochemistry category:
Site-Specific Inhibition of Translation Initiation via 2’-O-methylation
Leonie Brüne
Inhibition of VP2-mediated entry: a potential antiviral strategy to treat or prevent calicivirus disease
Orestis Savva
Inhibition of the gut ceramidase Asah2 decelerates the vertebrate ageing rate
Jeny Jose
Also in the cell biology category:
Combinatorial and Inducible CRISPRa/i Enables Canalized hiPSC Forward Programming and Iterative Refinement via Single-Cell Genomics
Cell-ID
Developmental conversion of the nucleolus into an RNA Polymerase II transcriptional platform in Drosophila spermatocytes
Panagiotis Giannios
Cell position is more important than cell shape or age for the acquisition of cell identity in the brown alga Ectocarpus
Urvashi Goswami
Also in the neuroscience category:
Behavioral characteristics of an extremely old rhesus macaque in a zoo: Dementia-like symptoms and implications for quality of life of geriatric animals
Stefan Friedrich Wirth
EBV reprograms autoreactive anti-CNS B cells as antigen presenting cells in multiple sclerosis
Léa Bastien et al.
The Endocannabinoid System’s Contribution to Placebo Analgesia
Thomas Nicodemo Arrieta et al.
preLists in the biochemistry category:
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
Peer Review in Biomedical Sciences
Communication of scientific knowledge has changed dramatically in recent decades and the public perception of scientific discoveries depends on the peer review process of articles published in scientific journals. Preprints are key vehicles for the dissemination of scientific discoveries, but they are still not properly recognized by the scientific community since peer review is very limited. On the other hand, peer review is very heterogeneous and a fundamental aspect to improve it is to train young scientists on how to think critically and how to evaluate scientific knowledge in a professional way. Thus, this course aims to: i) train students on how to perform peer review of scientific manuscripts in a professional manner; ii) develop students' critical thinking; iii) contribute to the appreciation of preprints as important vehicles for the dissemination of scientific knowledge without restrictions; iv) contribute to the development of students' curricula, as their opinions will be published and indexed on the preLights platform. The evaluations will be based on qualitative analyses of the oral presentations of preprints in the field of biomedical sciences deposited in the bioRxiv server, of the critical reports written by the students, as well as of the participation of the students during the preprints discussions.
| List by | Marcus Oliveira et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
20th “Genetics Workshops in Hungary”, Szeged (25th, September)
In this annual conference, Hungarian geneticists, biochemists and biotechnologists presented their works. Link: http://group.szbk.u-szeged.hu/minikonf/archive/prg2021.pdf
| List by | Nándor Lipták |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Cellular metabolism
A curated list of preprints related to cellular metabolism at Biorxiv by Pablo Ranea Robles from the Prelights community. Special interest on lipid metabolism, peroxisomes and mitochondria.
| List by | Pablo Ranea Robles |
MitoList
This list of preprints is focused on work expanding our knowledge on mitochondria in any organism, tissue or cell type, from the normal biology to the pathology.
| List by | Sandra Franco Iborra |
Also in the cell biology category:
Developmental regulation: molecular and ecological niches
This conference was held at the Station Biologique de Roscoff (France) and brought together researchers exploring how diverse niche environments shape developmental processes across scales. Spanning topics from ecological and metabolic influences to signalling networks, mechanics and gene regulation, the meeting highlighted the interplay between intrinsic and extrinsic factors in controlling cell fate and tissue organisation. This preList gathers preprints discussed by speakers and poster presenters during the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
SciELO preprints – From 2025 onwards
SciELO has become a cornerstone of open, multilingual scholarly communication across Latin America. Its preprint server, SciELO preprints, is expanding the global reach of preprinted research from the region (for more information, see our interview with Carolina Tanigushi). This preList brings together biological, English language SciELO preprints to help readers discover emerging work from the Global South. By highlighting these preprints in one place, we aim to support visibility, encourage early feedback, and showcase the vibrant research communities contributing to SciELO’s open science ecosystem.
| List by | Carolina Tanigushi |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
September in preprints – Cell biology edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading list. This month, categories include: (1) Cell organelles and organisation, (2) Cell signalling and mechanosensing, (3) Cell metabolism, (4) Cell cycle and division, (5) Cell migration
| List by | Sristilekha Nath et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
June in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell organelles and organisation (2) Cell signaling and mechanosensation (3) Genetics/gene expression (4) Biochemistry (5) Cytoskeleton
| List by | Barbora Knotkova et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
Keystone Symposium – Metabolic and Nutritional Control of Development and Cell Fate
This preList contains preprints discussed during the Metabolic and Nutritional Control of Development and Cell Fate Keystone Symposia. This conference was organized by Lydia Finley and Ralph J. DeBerardinis and held in the Wylie Center and Tupper Manor at Endicott College, Beverly, MA, United States from May 7th to 9th 2025. This meeting marked the first in-person gathering of leading researchers exploring how metabolism influences development, including processes like cell fate, tissue patterning, and organ function, through nutrient availability and metabolic regulation. By integrating modern metabolic tools with genetic and epidemiological insights across model organisms, this event highlighted key mechanisms and identified open questions to advance the emerging field of developmental metabolism.
| List by | Virginia Savy, Martin Estermann |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
March in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cancer biology 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics and genomics 6) other
| List by | Girish Kale et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
February in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry and cell metabolism 2) cell organelles and organisation 3) cell signalling, migration and mechanosensing
| List by | Barbora Knotkova et al. |
Community-driven preList – Immunology
In this community-driven preList, a group of preLighters, with expertise in different areas of immunology have worked together to create this preprint reading list.
| List by | Felipe Del Valle Batalla et al. |
January in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell migration 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Barbora Knotkova et al. |
December in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) cell cycle and division 2) cell migration and cytoskeleton 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) genetics/gene expression
| List by | Matthew Davies et al. |
November in preprints – the CellBio edition
This is the first community-driven preList! A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. Categories include: 1) cancer cell biology 2) cell cycle and division 3) cell migration and cytoskeleton 4) cell organelles and organisation 5) cell signalling and mechanosensing 6) genetics/gene expression
| List by | Felipe Del Valle Batalla et al. |
BSCB-Biochemical Society 2024 Cell Migration meeting
This preList features preprints that were discussed and presented during the BSCB-Biochemical Society 2024 Cell Migration meeting in Birmingham, UK in April 2024. Kindly put together by Sara Morais da Silva, Reviews Editor at Journal of Cell Science.
| List by | Reinier Prosee |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
preLights peer support – preprints of interest
This is a preprint repository to organise the preprints and preLights covered through the 'preLights peer support' initiative.
| List by | preLights peer support |
The Society for Developmental Biology 82nd Annual Meeting
This preList is made up of the preprints discussed during the Society for Developmental Biology 82nd Annual Meeting that took place in Chicago in July 2023.
| List by | Joyce Yu, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
9th International Symposium on the Biology of Vertebrate Sex Determination
This preList contains preprints discussed during the 9th International Symposium on the Biology of Vertebrate Sex Determination. This conference was held in Kona, Hawaii from April 17th to 21st 2023.
| List by | Martin Estermann |
Alumni picks – preLights 5th Birthday
This preList contains preprints that were picked and highlighted by preLights Alumni - an initiative that was set up to mark preLights 5th birthday. More entries will follow throughout February and March 2023.
| List by | Sergio Menchero et al. |
CellBio 2022 – An ASCB/EMBO Meeting
This preLists features preprints that were discussed and presented during the CellBio 2022 meeting in Washington, DC in December 2022.
| List by | Nadja Hümpfer et al. |
Fibroblasts
The advances in fibroblast biology preList explores the recent discoveries and preprints of the fibroblast world. Get ready to immerse yourself with this list created for fibroblasts aficionados and lovers, and beyond. Here, my goal is to include preprints of fibroblast biology, heterogeneity, fate, extracellular matrix, behavior, topography, single-cell atlases, spatial transcriptomics, and their matrix!
| List by | Osvaldo Contreras |
EMBL Synthetic Morphogenesis: From Gene Circuits to Tissue Architecture (2021)
A list of preprints mentioned at the #EESmorphoG virtual meeting in 2021.
| List by | Alex Eve |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
Planar Cell Polarity – PCP
This preList contains preprints about the latest findings on Planar Cell Polarity (PCP) in various model organisms at the molecular, cellular and tissue levels.
| List by | Ana Dorrego-Rivas |
BioMalPar XVI: Biology and Pathology of the Malaria Parasite
[under construction] Preprints presented at the (fully virtual) EMBL BioMalPar XVI, 17-18 May 2020 #emblmalaria
| List by | Dey Lab, Samantha Seah |
1
Cell Polarity
Recent research from the field of cell polarity is summarized in this list of preprints. It comprises of studies focusing on various forms of cell polarity ranging from epithelial polarity, planar cell polarity to front-to-rear polarity.
| List by | Yamini Ravichandran |
TAGC 2020
Preprints recently presented at the virtual Allied Genetics Conference, April 22-26, 2020. #TAGC20
| List by | Maiko Kitaoka et al. |
3D Gastruloids
A curated list of preprints related to Gastruloids (in vitro models of early development obtained by 3D aggregation of embryonic cells). Updated until July 2021.
| List by | Paul Gerald L. Sanchez and Stefano Vianello |
ECFG15 – Fungal biology
Preprints presented at 15th European Conference on Fungal Genetics 17-20 February 2020 Rome
| List by | Hiral Shah |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
EMBL Seeing is Believing – Imaging the Molecular Processes of Life
Preprints discussed at the 2019 edition of Seeing is Believing, at EMBL Heidelberg from the 9th-12th October 2019
| List by | Dey Lab |
Autophagy
Preprints on autophagy and lysosomal degradation and its role in neurodegeneration and disease. Includes molecular mechanisms, upstream signalling and regulation as well as studies on pharmaceutical interventions to upregulate the process.
| List by | Sandra Malmgren Hill |
Lung Disease and Regeneration
This preprint list compiles highlights from the field of lung biology.
| List by | Rob Hynds |
Cellular metabolism
A curated list of preprints related to cellular metabolism at Biorxiv by Pablo Ranea Robles from the Prelights community. Special interest on lipid metabolism, peroxisomes and mitochondria.
| List by | Pablo Ranea Robles |
BSCB/BSDB Annual Meeting 2019
Preprints presented at the BSCB/BSDB Annual Meeting 2019
| List by | Dey Lab |
Biophysical Society Annual Meeting 2019
Few of the preprints that were discussed in the recent BPS annual meeting at Baltimore, USA
| List by | Joseph Jose Thottacherry |
ASCB/EMBO Annual Meeting 2018
This list relates to preprints that were discussed at the recent ASCB conference.
| List by | Dey Lab, Amanda Haage |
Also in the neuroscience category:
preLighters’ choice – Handpicked DevBio preprints
preLighters with expertise across developmental and stem cell biology have nominated a few developmental biology (and related) preprints they’re excited about and explain in a few paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Theodora Stougiannou et al. |
BSDB Spring Meeting: Molecules to Morphogenesis
The British Society for Developmental Biology (BSDB) Spring Meeting Molecules to Morphogenesis was held from 23–26 March 2026 at the University of Warwick (UK). This meeting brought together a vibrant community of researchers to discuss how molecular mechanisms are integrated across scales to drive morphogenesis, spanning diverse model systems and approaches. This preList contains preprints by presenters from the talk and poster sessions at the meeting. Please do get in touch at preLights@biologists.com if you notice any relevant preprints that we may have missed.
| List by | Ingrid Tsang |
Keystone Symposium on Stem Cell Models in Embryology 2026
The Keystone Symposium on Stem Cell Models in Embryology, 2026, was organised by Jun Wu (UT Southwestern), Jianping Fu (University of Michigan) and Miki Ebisuya (TU Dresden) and held at Asilomar Conference Grounds in California (US). The meeting discussed recent advances made in establishing stem-cell-based embryo models, what fundamental insights into developmental processes have been gleaned from them, as well as how they are beginning to be applied more widely. This prelist contains preprints by presenters at the talk and poster sessions at the conference, which our Reviews Editor in attendance spotted. Please do reach out to preLights@biologists.com if you notice any that we’ve missed.
| List by | Ingrid Tsang |
November in preprints – DevBio & Stem cell biology
preLighters with expertise across developmental and stem cell biology have nominated a few developmental and stem cell biology (and related) preprints posted in November they’re excited about and explain in a single paragraph why. Concise preprint highlights, prepared by the preLighter community – a quick way to spot upcoming trends, new methods and fresh ideas.
| List by | Aline Grata et al. |
October in preprints – DevBio & Stem cell biology
Each month, preLighters with expertise across developmental and stem cell biology nominate a few recent developmental and stem cell biology (and related) preprints they’re excited about and explain in a single paragraph why. Short, snappy picks from working scientists — a quick way to spot fresh ideas, bold methods and papers worth reading in full. These preprints can all be found in the October preprint list published on the Node.
| List by | Deevitha Balasubramanian et al. |
October in preprints – Cell biology edition
Different preLighters, with expertise across cell biology, have worked together to create this preprint reading list for researchers with an interest in cell biology. This month, most picks fall under (1) Cell organelles and organisation, followed by (2) Mechanosignaling and mechanotransduction, (3) Cell cycle and division and (4) Cell migration
| List by | Matthew Davies et al. |
July in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: (1) Cell Signalling and Mechanosensing (2) Cell Cycle and Division (3) Cell Migration and Cytoskeleton (4) Cancer Biology (5) Cell Organelles and Organisation
| List by | Girish Kale et al. |
May in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) Biochemistry/metabolism 2) Cancer cell Biology 3) Cell adhesion, migration and cytoskeleton 4) Cell organelles and organisation 5) Cell signalling and 6) Genetics
| List by | Barbora Knotkova et al. |
April in preprints – the CellBio edition
A group of preLighters, with expertise in different areas of cell biology, have worked together to create this preprint reading lists for researchers with an interest in cell biology. This month, categories include: 1) biochemistry/metabolism 2) cell cycle and division 3) cell organelles and organisation 4) cell signalling and mechanosensing 5) (epi)genetics
| List by | Vibha SINGH et al. |
Biologists @ 100 conference preList
This preList aims to capture all preprints being discussed at the Biologists @100 conference in Liverpool, UK, either as part of the poster sessions or the (flash/short/full-length) talks.
| List by | Reinier Prosee, Jonathan Townson |
2024 Hypothalamus GRC
This 2024 Hypothalamus GRC (Gordon Research Conference) preList offers an overview of cutting-edge research focused on the hypothalamus, a critical brain region involved in regulating homeostasis, behavior, and neuroendocrine functions. The studies included cover a range of topics, including neural circuits, molecular mechanisms, and the role of the hypothalamus in health and disease. This collection highlights some of the latest advances in understanding hypothalamic function, with potential implications for treating disorders such as obesity, stress, and metabolic diseases.
| List by | Nathalie Krauth |
‘In preprints’ from Development 2022-2023
A list of the preprints featured in Development's 'In preprints' articles between 2022-2023
| List by | Alex Eve, Katherine Brown |
CSHL 87th Symposium: Stem Cells
Preprints mentioned by speakers at the #CSHLsymp23
| List by | Alex Eve |
Journal of Cell Science meeting ‘Imaging Cell Dynamics’
This preList highlights the preprints discussed at the JCS meeting 'Imaging Cell Dynamics'. The meeting was held from 14 - 17 May 2023 in Lisbon, Portugal and was organised by Erika Holzbaur, Jennifer Lippincott-Schwartz, Rob Parton and Michael Way.
| List by | Helen Zenner |
FENS 2020
A collection of preprints presented during the virtual meeting of the Federation of European Neuroscience Societies (FENS) in 2020
| List by | Ana Dorrego-Rivas |
ASCB EMBO Annual Meeting 2019
A collection of preprints presented at the 2019 ASCB EMBO Meeting in Washington, DC (December 7-11)
| List by | Madhuja Samaddar et al. |
SDB 78th Annual Meeting 2019
A curation of the preprints presented at the SDB meeting in Boston, July 26-30 2019. The preList will be updated throughout the duration of the meeting.
| List by | Alex Eve |
Autophagy
Preprints on autophagy and lysosomal degradation and its role in neurodegeneration and disease. Includes molecular mechanisms, upstream signalling and regulation as well as studies on pharmaceutical interventions to upregulate the process.
| List by | Sandra Malmgren Hill |
Young Embryologist Network Conference 2019
Preprints presented at the Young Embryologist Network 2019 conference, 13 May, The Francis Crick Institute, London
| List by | Alex Eve |
5 years
Patrick Ernst
Hi, first of all thank you for highlighting this, I’m always glad when people take an interest in my work. Sorry to not address your questions sooner – I only came across this now. I hope you find these answers useful, feel free to reach out if you have any more questions. As to your questions:
1) How do you know the ChR2 is definitely at the IMM?
I know that there are some methods to isolate the IMM, but as far as I know those can be difficult. By far THE biggest piece of evidence we have that ChR2 is in the IMM is the fact that it works as intended. We know from fluorescent imaging and western blotting that ChR2-eYFP expression is colocalized with mitochondria, meaning it is either in/attached to the outer membrane (OMM), in the inter-membrane space, in the IMM, or in the mitochondrial matrix. If ChR2 were expressed in the OMM, inter-membrane space, or matrix (or improperly oriented in the IMM), activation via blue light stimulation would have little to no effect on the membrane potential of the IMM. It feels a bit cheap to just say “because it works” as an explanation, but the statistical likelihood of it working and NOT being in the IMM is low enough that it’s a satisfactory answer for me.
2) Is it possible to block or mutate the ChR2 channel to determine whether it is definitely a transfer of protons through this channel causing depolarisation?
A lot of work has been done to mutate ChR2 in order to change its physical properties (e.g. redshifting the absorption spectrum, increasing photocurrent), it has been shown that mutations can affect ion sensitivity/selectability. Cho et al were able to mutate ChR2 to significantly reduce proton conductance by a factor of ~20, so this is definitely within the realm of possibility.
However, when you take into consideration the ion-specific conductivities of ChR2 as well as comparing the intra- and extra-mitochondrial concentrations of those common ions, it follows that the majority of the depolarizing ion flow is due to protons. Schneider et al looked at ion-specific conduction of ChR2 and found that under (mostly) physiological conditions the majority (~65% by my estimate, Figure 6A) of inward current (both initial and steady-state) was due to protons, and that Ca2+ was somewhat suppressed due to competition with protons. It’s not a perfect comparison, as the ion gradients across the cell membrane will differ from those across the IMM, but it does still suggest higher conductivity of protons over other ions and that higher proton current may inhibit conductivity of other ions. Further, it is well-established that the membrane potential of the IMM is predominantly due to the strong proton gradient across the IMM – the ETC utilizes this in ATP production. It follows that taking an ion channel that preferentially conducts H+ (in circumstances where there is a low H+ gradient and large Na+ or Ca2+ gradients) and then placing it in an environment with a very large H+ gradient and much lower Na+ and Ca2+ gradients would elicit conduction almost entirely made up of protons.
Additionally, while not shown in the manuscript we did investigate possible mitochondrial Ca2+ influx (at the request of a reviewer) and found no significant change to intra-mitochondrial Ca2+, suggesting negligible Ca2+ flux through mitochondrial ChR2 activation.
3) Can you speculate as to what mechanisms induce this protection of cell viability when mitochondria are preconditioned? Has anyone looked at this?
I personally don’t know, to such an extent that it would probably be irresponsible of me to guess anything specific. The core concept is straight-forward enough – submit cells to sublethal stress, and they will adapt against that stress, making them more resilient in the near future. But past that my understanding of it is fairly limited. Luckily mitochondrial preconditioning is something that people have been using and studying for years now, typically in the context of ischemic preconditioning in neurons. Correia et al. provide a review of the topic and detail some potential mechanisms involved. Note, there may be some differences in our case, due to the difference in stressors/mechanisms between ischemic preconditioning and our photostimulation.
I recently graduated and have since left that lab/project but I know they’re currently looking into the specific mechanisms behind the cytoprotective effects we’ve seen, hopefully they find something compelling! Since publishing this paper, we have tested the effects of optogenetic mitochondrial preconditioning in the context of other more common stressors and it did have a protective effect against those as well, so the underlying mechanisms may be more similar than I think.
References:
Y.K. Cho, D. Park, A. Yang, F. Chen, A.S. Chuong, N.C. Klapoetke, E.S. Boyden. Multimensional screening yields channelrhodopsin variants having improved photocurrent and order-of-magnitude reductions in calcium and proton currents. J Bio Chem. 2019;294(11):P3806-3821. doi:10.1074/jbc.RA118.006996
F. Schneider, D. Gradmann, P. Hegemann. Ion selectivity and competition in channelrhodopsins. Biophys J. 2013;105(1):91-100. doi:10.1016/j.bpj.2013.05.042
S.C. Correia, C. Carvalho, S. Cardoso, R.X. Santos, M.S. Santos, C.R. Oliveira, G. Perry, X. Zhu, M.A. Smith, P.I. Moreira. Mitochondrial preconditioning: a potential neuroprotective strategy. Front Aging Neurosci. 2010;2:183. doi:10.3389/fnagi.2010.00138